All published articles of this journal are available on ScienceDirect.

Structural Modifications of Curcumin toward Clinical Applications
Authors Info & Affiliations
Abstract
Curcumin has attracted significant attention for its therapeutic potential across a range of diseases, including cancer, inflammation, and neurodegenerative disorders. However, its clinical efficacy is limited by poor bioavailability, instability under physiological conditions, and a lack of specificity. This review explores chemical strategies to address these limitations, focusing on enhancing curcumin’s bioavailability, metabolic stability, and target specificity, particularly against DYRK-2. It examines diverse structural modifications of curcumin, including reducing the diketo group to a monoketo form, substituting the diketo group with heterocyclic rings, and other targeted modifications aimed at overcoming curcumin's pharmacokinetic challenges. Recent advancements, including the development of curcumin analogues with improved pharmacokinetic profiles, are highlighted, alongside an evaluation of their impact on bioactivity and therapeutic efficacy. Additionally, persistent challenges, such as toxicity, selectivity, and reactivity with biomolecules like glutathione, are identified. By incorporating current findings and future directions, this review highlights the need for continued innovation in curcumin analogue development to realise its clinical potential.
1. INTRODUCTION
Curcumin, often referred to as the “golden spice,” has captivated the attention of the scientific community, appearing in more than 46,000 publications since its initial identification in 1815. Its structure was elucidated almost a century later, and its antimicrobial activity was first reported in 1949, paving the way for further exploration of its pharmacological potential. Over the years, curcumin has been recognised for its therapeutic properties across a wide range of diseases, including cancer, through phenotypic assays [1]. As a component of turmeric, which has been used for centuries in traditional Indian and Chinese medicine, curcumin is a bioactive molecule of interest to modern drug discovery. Historical records trace the medicinal use of turmeric, or haridra, to Hindu texts from the tenth century [2].
Despite curcumin’s promising performance in preclinical studies and the initiation of approximately 343 clinical trials (ClinicalTrials.gov), its translational potential remains elusive. Clinical studies have demonstrated symptomatic improvements and histological changes in cancer patients treated with curcumin in various formulations, such as capsules, ointments, and extracts. In multiple myeloma, high oral doses of curcumin have been associated with decreased cancer-related protein expression and improved clinical markers. Similar results have been observed in colorectal cancer, where curcumin reduced inflammatory markers and polyp size and enhanced apoptosis. Yet curcumin has demonstrated limited efficacy in advanced pancreatic cancer [3].
The binding of curcumin to proteins, such as transthyretin, which regulates thyroid hormone and retinol transport, and dual-specificity tyrosine-regulated kinase 2 (DYRK2), validates its relevance as a pharmacological target [4, 5]. However, despite these promising interactions, curcumin's pharmacokinetic profile, including poor oral bioavailability, rapid metabolism, and low physiological stability, is an issue that needs to be addressed before it can be considered a “magic bullet” for cancer therapy.
The chemical structure of curcumin, characterised by hydrophobic aromatic rings and an α, β-unsaturated β-diketo chain, is responsible for its lipophilic nature and solubility profile. The presence of this chain, while contributing to the molecule's flexibility and ability to interact with multiple targets as a Michael acceptor [6, 7], is also responsible for its rapid degradation at physiological pH. The compound exists in its enol tautomeric form at physiological pH, with a reported half-life of less than ten minutes in humans, undergoing rapid autoxidation to form a bicyclopentadione product [8-11]. The metabolites of curcumin, such as ferulic acid, vanillin, and bicyclopentadione, exhibit significantly reduced bioactivity compared to the parent compound [12].
Numerous efforts have been made to modify curcumin’s chemical structure, particularly the unsaturated β-diketo chain, to enhance its bioavailability, metabolic stability, and anticancer activity. Unfortunately, these attempts have largely been met with limited success, underscoring the need for continued research to optimise the pharmacokinetic profile before pursuing further mechanistic studies to achieve translational success. This review critically assesses synthetic modifications to curcumin to date, evaluating their impact on improving its pharmacokinetics and anticancer efficacy while addressing the challenges that have hindered its clinical translation.
2. CHEMICAL MODIFICATIONS ON CURCUMIN – CURCUMIN ANALOGUES
2.1. Reduced Analogues of Curcumin
The analogues of curcumin, such as tetrahydrocurcumin and hexahydrocurcumin, have been the focus of several studies due to their improved chemical stability at physiological pH. Unlike curcumin, these analogues lack the unsaturated β-diketo chain, which contributes to curcumin's instability. Holder et al [13] identified glucuronides of tetrahydrocurcumin and hexahydrocurcumin. These compounds have been evaluated for various pharmacological properties, including anti-inflammatory, antioxidant, anticancer, hepatoprotective, antidiabetic, and photoprotective activities. Despite a broad bioactive profile, the low aqueous solubility of tetrahydrocurcumin limits its bioavailability [14, 15].
Hexahydrocurcumin, another analogue of curcumin, has shown better bioavailability in Caco2 permeability assays [16], and its anticancer, antioxidant, anti-inflammatory, and vasorelaxation activities have been identified [17]. Studies have shown that reduced curcumin analogues exhibit higher kinetic solubility than curcumin. However, tetrahydrocurcumin has lower metabolic stability compared to both hexahydrocurcumin and octahydrocurcumin in in vitro assays [18].
Dihydrocurcumin, another compound obtained from the reduction of curcumin, has demonstrated activity against human and rat 17β-HSD1 enzymes. It was found to inhibit oestradiol secretion in human BeWo cells at concentrations of ≥5–10 μM [19]. However, like other reduced analogues, dihydrocurcumin’s clinical utility is limited by poor bioavailability.
The number of publications evidences the interest in these reduced analogues. Over 400 articles in SCOPUS-indexed journals on tetrahydrocurcumin and more than 80 on hexahydrocurcumin have been published, highlighting their therapeutic potential and the need for improved formulations to address their poor pharmacokinetic profiles.
2.2. Monocarbonyl Analogues of Curcumin
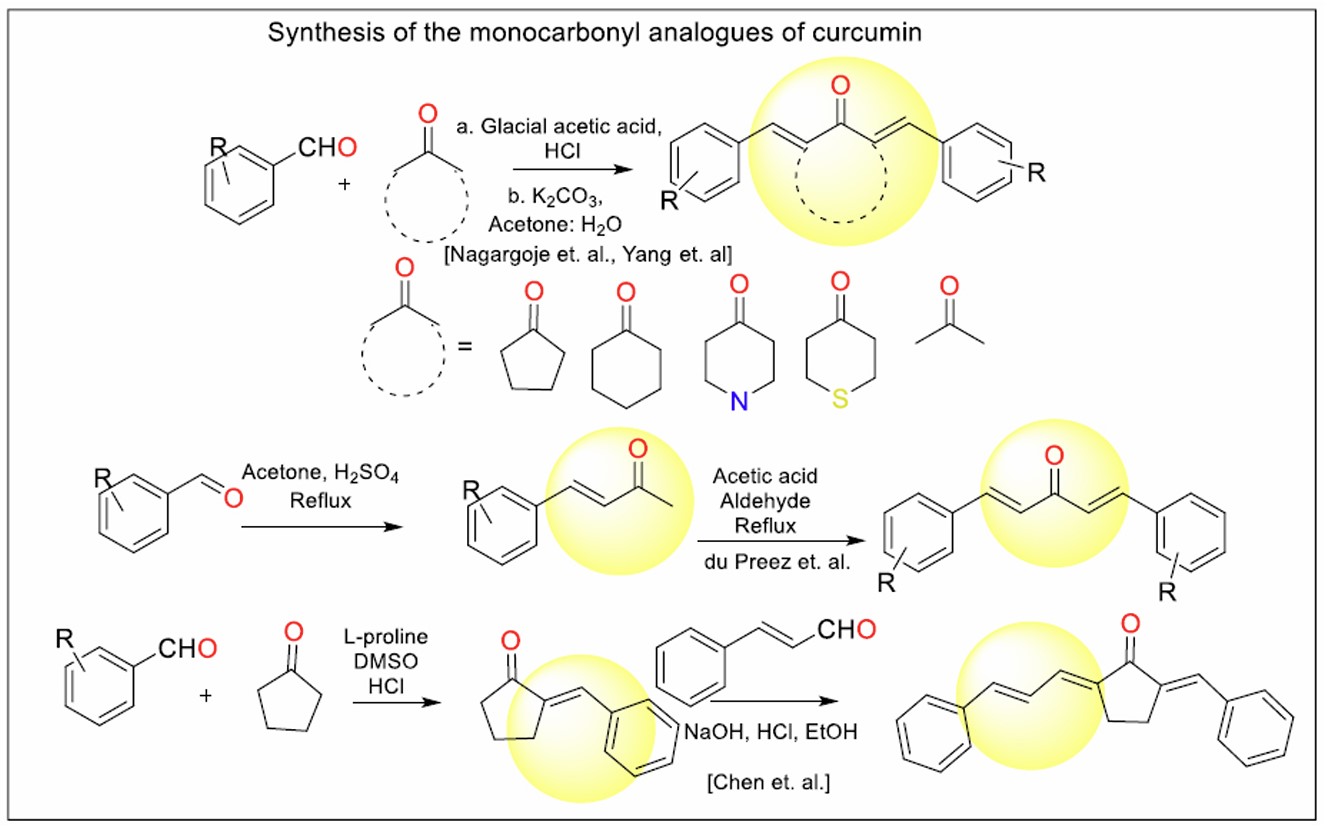
To address the instability associated with curcumin’s β-diketo chain, researchers have explored monocarbonyl analogues, which replace the β-diketo group with a more stable heterocyclic moiety. These compounds have been discussed here as piperidone analogues (Fig. 1, series 1a-1h), straight chain analogues (Fig. 2, series 2a-2j), cyclopentanone analogues (Fig. 3, series 3a-3h), and cyclohexanone analogues (Fig. 4, Series 5a – 5e)).
Youssef et al. [20] synthesised analogues in which the β-diketo group was replaced with various rings, including piperidone, cycloheptanone, cyclopentane, and cyclohexane. These modifications were aimed at increasing physiological stability while enhancing antioxidant and anticancer activities. However, removing or replacing methoxy groups with ethoxy groups diminished antioxidant potency. Additionally, the presence of a p-hydroxylic group and an electron-releasing group ortho to the phenolic hydroxyl was found to be crucial for maintaining antioxidant activity. Chen et al. [21] identified the brominated analogue NL04 (Fig. 1a), which exhibited pain-relieving properties in bone cancer models via NLRP3 and IL-1β inhibition.
Adams et al. [22] synthesised a series of monocarbonyl analogues, which feature a shortened five-carbon chain between the aromatic rings instead of curcumin’s seven-carbon chain. Among monocarbonyl curcumin analogues, EF-24 (Fig. 1b), a piperidone analogue, was found to have higher potency than curcumin with respect to anticancer and anti-angiogenic properties. Krasinski et al. [23] reported that EF-24 affects the NF-κB pathway, while Reid et al [24] identified its high oral bioavailability and rapid clearance in rats. Despite its promising activity, formulation is a challenge. Agashe et al. [25] reported a liposome-encapsulated EF-24 formulation with a short half-life. EF-31 (Fig. 1c), a more potent analogue of EF-24, features phenyl rings replaced with piperidine rings, further enhancing its biological activity [26].
EF-31 (Fig. 1c), a more potent analogue of EF-24, has been found to induce anticancer effects through DNA methyltransferase downregulation in pancreatic cancer cells and increases PON1 activity in the U87MG glioblastoma cell line [27]. Further studies on EF-24 analogues, such as UBS109, demonstrated additional anticancer activity, including migration inhibition in HeLa and SiHa cervical cancer cells by influencing matrix metalloproteinases and the phosphorylation of the p38 signalling pathway [28-31].
In addition to EF-24, other monocarbonyl analogues have been studied for their potential therapeutic applications. Malave et al. [32] synthesised N-Boc-protected analogues of piperidone chalcones, identifying four with potent selectivity for colorectal cancer cells. Ghosh et al. [33] explored structural modifications of EF-24 and observed that N-acylation of EF-24 did not enhance its potency. However, when fluorine was introduced at the third, or both the third and fourth positions, of the benzylidene rings, and a trifluoromethyl group was added at the fourth position, the resulting acrolyl analogues exhibited a significantly improved activity profile compared to EF-24. These modifications suggest that substituting with electron-withdrawing groups, such as fluorine and trifluoromethyl, may enhance the pharmacological potential of curcumin analogues.
Zhuang et al. [34] reported ACE inhibitory activity for the curcumin analogue – UBS109 (Fig. 1d), with better bioavailability than curcumin. Further evaluations by Razali et al. followed. [35], who tested the anticancer effects of two piperidone analogues, FLDP-5 and FLDP-8, in glioblastoma cells. D’Arcy et al. [36] identified b-AP15 (Fig. 1e) as a proteasome inhibitor that targets deubiquitinating enzymes in the 19S subunit of the proteasome.
Nishimura et al. [37] evaluated NC2603 (Fig. 1f), another monocarbonyl analogue of curcumin, for its selective cytotoxicity activity against MCF-7 breast cancer cells (IC50 = 5.6 μM). Transcriptomic analysis of cells treated with NC2603 showed upregulation of CDKN1A, potentially via ESR1 or GADD45A. NC2603 was also shown to inhibit cell migration in BT-20 breast cancer cells (IC50=3.5 µΜ), possibly through EGR3 regulation [38].
Selvendiran et al. [39] reported HO-3867 (Fig. 1h), a difluoro monocarbonyl curcumin analogue substituted with N-hydroxy proline. The compound showed improved cell permeability as compared to curcumin [40]. The anticancer potential of HO-3867 has been evaluated against various cancers, including osteosarcoma [41] and hepatocellular carcinoma [42]. It has been shown to inhibit arterial smooth muscle proliferation and arterial stenosis [43]. The compound's effect on the JNK signalling pathway has also been explored, highlighting its effect on multiple pathways.
Liu et al. [44] synthesised a dibenzylidene piperidone analogue of curcumin, with trifluoromethyl groups at the third position of the benzylidene rings. This compound showed potent and selective anticancer activity against NCI-H460 lung cancer cells by inducing reactive oxygen species (ROS) generation. However, other structural modifications, including replacing the piperidone ring with a cyclohexane ring or substituting the piperidone with N-methyl-piperidone, yielded less potent compounds than the lead molecule identified in this study, underscoring the importance of specific structural features for anticancer activity.
Various research groups have continued to explore piperidone analogues of curcumin, testing them for a wide range of pharmacological activities. These analogues have been found to exhibit α-glucosidase inhibition [45], aldose reductase inhibition [46], anticancer properties [47-49] and anti-inflammatory activity [50], indicating their broad therapeutic potential. The structures of the most potent piperidone analogues of curcumin showing anticancer activity (Fig. 1g and i) [51, 52], antifungal activity (Fig. 1m and j) [53, 54], anti-inflammatory activity (1k) [55] and antitubercular activity [56] have been represented in Fig. (1).
Wang et al. [57] further expanded on this work by identifying a monocarbonyl curcumin analogue that induced apoptosis in H460 human lung cancer cells by upregulating C/EBP-homologous protein expression, a key regulator of cell stress response pathways. In related studies, Xiao et al. [58] discovered B63 (Fig. 2a), a monocarbonyl curcumin analogue where the bismethoxyphenyl groups were connected by a penta-1,4-dien-3-one chain. B63 demonstrated apoptotic activity against human non-small cell lung cancer, highlighting the importance of chain length and substitution patterns in improving the efficacy of curcumin analogues.
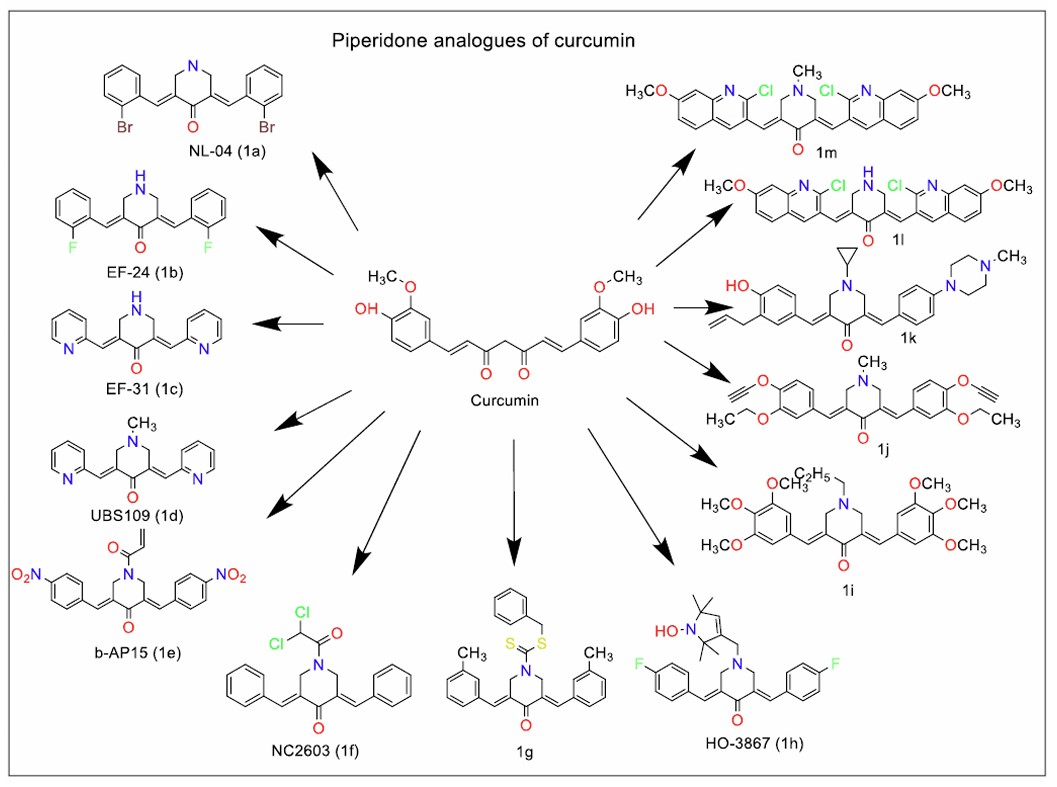
Piperidone analogues of curcumin (Series 1).
Liu et al. [59] identified that B82, a bromo-substituted analogue of B63, which effectively inhibited H460 tumour growth in in vivo experiments, underlining the anticancer potential of monocarbonyl curcumin analogues. Further, Luo et al. [60] and Tan et al. [61] reported dibenzylidene thiopyranone derivatives of curcumin, more resistant to hydrolysis. These compounds were more potent than curcumin and triggered apoptosis in leukemic cells by regulating endoplasmic reticulum stress signalling pathways. This mechanism represents a novel therapeutic approach for targeting cancer cells resistant to conventional chemotherapeutic agents.
Hutzen et al. [62] identified GO-Y030 (Fig. 2c), a straight chain monocarbonyl analogue of curcumin, that exhibited potent cytotoxicity against both breast cancer and pancreatic cancer cell lines. GO-Y030's anticancer efficacy was linked to the regulation of cancer cell proliferation by downregulating the transcription factor STAT3. Kudo et al. [63] investigated GO-Y030 and its analogue GO-Y078 (Fig. 2f), identifying that both compounds effectively targeted multiple oncogenic pathways, including NF-κB, PI3K/AKT, JAK/STAT3, and IRF4. These pathways play crucial roles in tumour cell survival and drug resistance, making these analogues promising candidates for further development.
The ability of GO-Y030 and its analogues to inhibit the ATP-binding cassette (ABC) transporter, a protein involved in cancer cell drug resistance mechanisms, was identified by Murakami et al. [64]. However, the bioavailability of GO-Y030 was limited by its poor water solubility. To address this issue, Yamakoshi et al. [65] synthesised GO-Y199, a prodrug of GO-Y030. This prodrug, modified with propargyl-polyethene glycol and 1-glycosylazide, exhibited significantly improved aqueous solubility at concentrations of 100 µM while maintaining anticancer activity comparable to that of the parent compound. These modifications open the door to analogues with a better pharmacokinetic profile, suitable for in vivo application. Extensive research continues to explore the in vitro and in vivo anticancer activities of these curcumin analogues, reinforcing their potential as therapeutic agents.
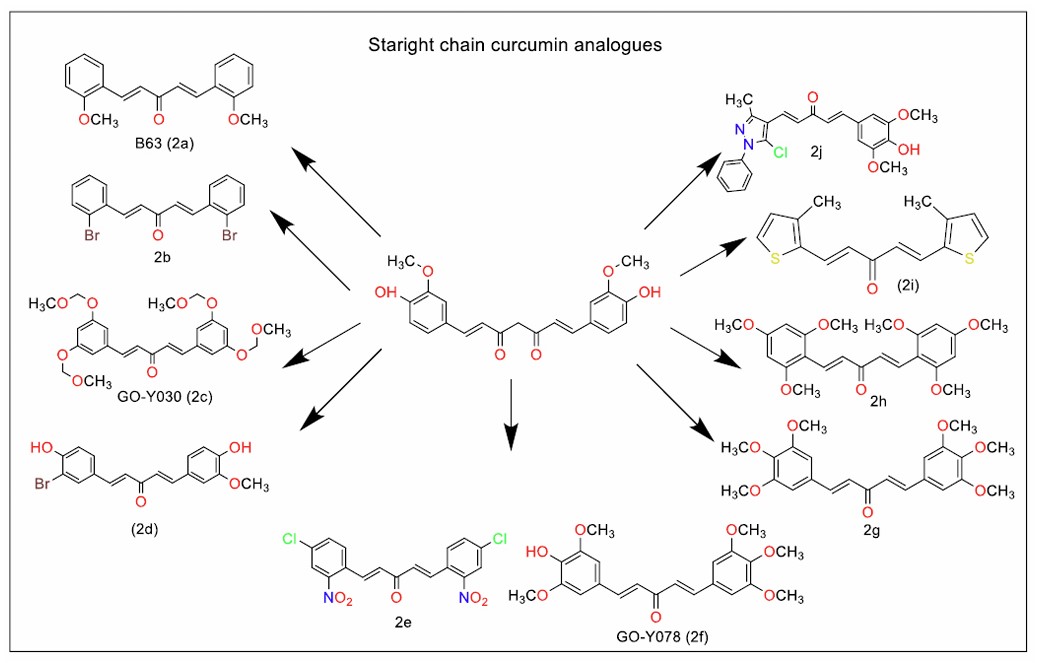
Straight chain analogues of curcumin.
Li et al. [66] identified two hexamethoxy-diarylpentanedienone analogues of curcumin (Fig. 2g and h), which were shown to induce G2/M phase arrest in NCI-H460 lung cancer cells, possibly through the induction of reactive oxygen species (ROS). By overwhelming the cell's redox buffering system, these analogues significantly disrupted cellular homeostasis, leading to cell cycle arrest. The structures of potent straight-chain monocarbonyl curcumin analogues with anticancer activity (Fig. 2b, d, and j) [67-69], antitubercular activity [70] have been included in Fig. (2).
Further expanding on the therapeutic potential of curcumin analogues, Liang et al. [71] synthesised dibenzylidenecyclopentanone analogues (Fig. 3f and g) and a mono-keto analogue, where the phenyl rings were replaced with thiophene (Fig. 2i). These compounds demonstrated potent anti-inflammatory properties by inhibiting the release of key inflammatory mediators, including TNF-α, IL-1β, IL-6, MCP-1, COX-2, PGES, iNOS, and p65 NF-κB mRNA. Notably, the analogue with a dipropyloxy dimethylamino group (Fig. 3d) showed enhanced water solubility when formulated as its hydrochloride salt, which improved its potential for clinical applications.
Chen et al. identified cyclopentanone curcumin analogues synthesised from cinnamaldehyde and demonstrated potential anticancer activity in vitro and in murine models. One compound (Fig. 3a) showed inhibitory activity against the SGC-7901 and AGS cell lines, with an IC50 value of less than 1 µM. Replacement of the hydroxyl group with halogens resulted in a complete lack of antiproliferative activity [72].
Zhao et al. [73] identified that diarylpenta-1,4-diene-3-one analogues of curcumin exhibited superior anti-inflammatory activity compared to both dibenzylidene cyclohexanone and dibenzylidene cyclopentanone analogues in LPS-stimulated RAW 264.7 macrophages. One particular analogue, C66 (Fig. 3b), a trifluoromethyl-substituted dibenzylidene cyclopentanone, was found to prevent renal injury in mice by inhibiting high glucose-induced inflammation and reducing macrophage infiltration [74].
Taking these findings further, Feng et al. [75] explored 34 asymmetric analogues of C66 (Fig. 3c and e) and demonstrated their potent in vivo anti-inflammatory effects in LPS-induced acute lung injury in rats. These analogues inhibited the expression of inflammatory mediators at the mRNA level and demonstrated cytoprotective effects, as evidenced by histopathological examination of the treated rats.
Hu et al. [76] identified that C66 reduced the disease activity index in a dextran sodium sulphate (DSS) induced IBD (inflammatory bowel disease) model in mice, showing promise for treating IBD. Mladenov et al. [77] reported the cardioprotective effect of C66 against oxidative damage in diabetic cardiac tissue, possibly through regulation of Nrf2 and JNK2 pathways. C66's improved bioavailability and metabolic stability over curcumin make it a valuable candidate for further clinical studies.
Ai et al. [78] synthesised monocarbonyl hybrids of curcumin and ligustrazine with selectivity against both drug-resistant and drug-sensitive lung cancer cells. These findings suggest that hybrid curcumin analogues may offer a novel approach to overcoming cancer drug resistance.
Tang et al. [79] identified a diaryl cyclopenta-1,4-diene-3-one analogue of curcumin (Fig. 3h) where the aryl rings were substituted with a fluorine atom and a trifluoromethyl group. This compound effectively inhibited extracellular matrix accumulation in models of chronic kidney disease, suggesting its potential to prevent kidney fibrosis and related pathologies.
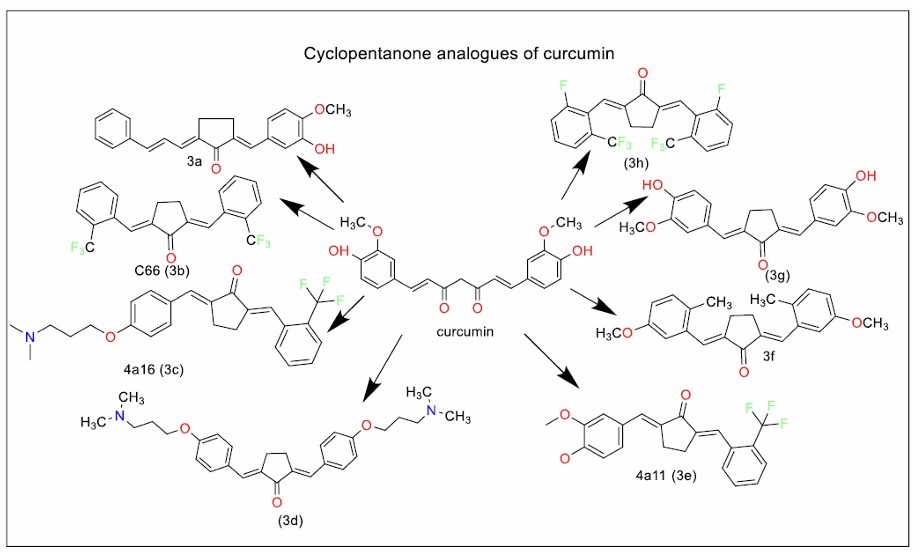
Cyclopentanone analogues of curcumin (Series 3).
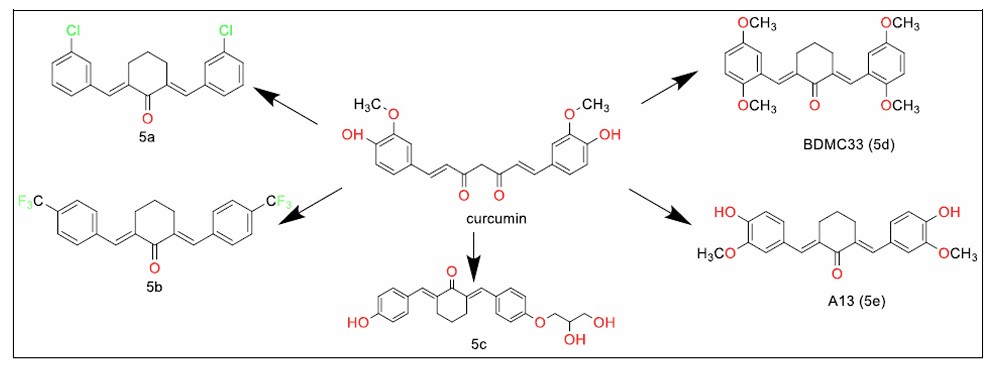
Cyclohexanone analogues of curcumin (Series 5).
Cyclohexanone analogues of curcumin (Fig. 4, 5a) with antiparasitic activity against Trichomonas vaginalis have been identified by Carapina de Silva et al. It reduced in vitro parasite viability by 70% as compared to the control group [80]. Liu B et al. identified the antiangiogenic activity of a cyclohexanone analogue of curcumin (Fig. 4, 5b) in in vitro, ex vivo, and in vivo studies, through modulation of NADH/NADPH oxidase-derived ROS [81]. This discovery could have implications for the control of tumour growth in cancer therapy.
A cyclohexanone analogue of curcumin with an IC50 of 6.27 µM against lung cancer cells - H460 and H1650, with a potential for the development of treatment options for lung cancer, was identified. Lee et al. [82] synthesised asymmetrically substituted dibenzylidene cyclohexanone and diarylpentan-1,4-dien-3-one curcumin analogues, discovering significant anti-inflammatory activity. Among them, the analogue BDMC33 (Fig. 4, 5d) showed superior activity, interfering with the NF-κB and MAPK signalling pathways in IFN-γ/LPS-stimulated macrophages. Additionally, BDMC33 disrupted LPS signalling by reducing the expression of CD-14 accessory molecules, thus inhibiting NO and TNF-α release in microglial cells. These properties position BDMC33 as a promising therapeutic agent for the treatment of chronic inflammatory conditions. In line with this, Mostafa et al. [83] identified BDMC33's anti-inflammatory potential in a model of bone morphogenetic protein-mediated inflammation and arthritis in zebrafish larvae.
Revalde et al. [84] identified A13 (Fig. 4, 5e), another monocarbonyl curcumin analogue, which showed significant potential in inhibiting the ATP-binding cassette (ABC) transporter. This activity is one of the crucial pathways in overcoming drug resistance in cancer cells. This positions A13 as a promising agent in combination therapies to combat multidrug-resistant cancers.
Aluwi et al. [85] substituted one of the phenyl rings of the asymmetric dibenzylidene cyclohexanone analogues with a furan ring, resulting in a compound with potent prostaglandin E2 inhibitory activity, extending the pharmacological relevance of curcumin analogues.
Qian et al. [86] identified a monoketo analogue of curcumin with a wide range of protective effects in a murine model of cardiac myopathy induced by a high-fat diet (HFD). The analogue exhibited anti-inflammatory, antioxidant, antifibrotic, and antiapoptotic effects, highlighting its therapeutic potential for cardiovascular diseases.
The growing body of research on monocarbonyl curcumin analogues, particularly GO-Y030 and EF-24, highlights their significant potential for cancer therapy.
2.3. Heterocyclic Analogues of Curcumin
Heterocyclic analogues of curcumin with potential as anti-inflammatory and anticancer therapies have been identified. Liu et al. [87] reported the anti-inflammatory activity of NSC 757929 (Fig. 5, 4b), a pyrazole analogue of curcumin substituted with a phenyl ring on the nitrogen atom of the pyrazole. This compound showed promising effects in murine models of Parkinson’s disease, underscoring the potential of curcumin analogues for treating neuroinflammatory conditions. Sahu et al. [88] synthesised pyrazole carboxamides (NSC 777947, Fig. 5, 4a), with anticancer activity against human lung cancer cells (IC50 ≈100 µM). Ahsan et al. [89] identified NSC 782199 (Fig. 5, 4f) as a potent anticancer agent across multiple human cancer cell lines. They synthesised various pyrazole analogues of curcumin, introducing modifications such as a carboxamide moiety on the pyrazole ring (Fig. 5, 4c), phenylmethanone groups (Fig. 5, 4d), and dihydropyrimidine derivatives (Fig. 6, 7a). The analogue with a phenylmethanone substitution on the pyrazole ring exhibited the highest anticancer potency, with a growth percentage of -28.71. The work was further expanded with the identification of three pyrazole analogues of curcumin - NSC 793120, NSC 782199, and NSC 782200 (Fig. 5, 4e, f, and h), which showed significant anticancer activity against multiple cancer cell lines. Substitution of the phenoxymethanone moiety with a phenoxyethanone moiety further enhanced potency, with over 80% growth inhibition.
Mohamed et al. [90] evaluated three pyrazole analogues of curcumin against cancer cells and confirmed their therapeutic potential for targeting epidermal growth factor receptor (EGFR). The analogue featuring 1-[[4-chlorophenyl] amino] propane-2-one on the pyrazole ring (Fig. 5, 4g) emerged as a potential candidate for further development. However, Ahsan et al. [91] identified the non-specific activity of these analogues, suggesting the need for additional modifications to improve selectivity against cancer cells.
Mari et al. [92] continued the structural optimisation of curcumin analogues with the synthesis of aminopyrimidine analogues (Fig. 7, 9a and b) with over 95% stability under physiological conditions. One compound, PY1 (Fig. 9a), was found to be equipotent to curcumin against prostate cancer cells. Another analogue, in which methyl groups replaced the methoxy groups of curcumin, showed better anticancer activity against HT-29 colorectal cancer cells, outperforming both curcumin and dimethoxy curcumin.
Feriotto et al. [93] focused on oxime, beta-enamine ketones, and isoxazole analogues of curcumin, targeting K562 leukaemia cells resistant to imatinib. These analogues were more stable compared to curcumin at physiological pH. The compounds containing bulky aryl rings on the enamine moiety showed greater stability than those substituted with smaller alkyl groups. Two isoxazole analogues, compounds 22 and 2 (Fig. 7, 9a and b), exhibited superior anticancer activity, with efficacy retained against imatinib-resistant cells. The absence of the methoxy group did not affect the potency of compound 22.
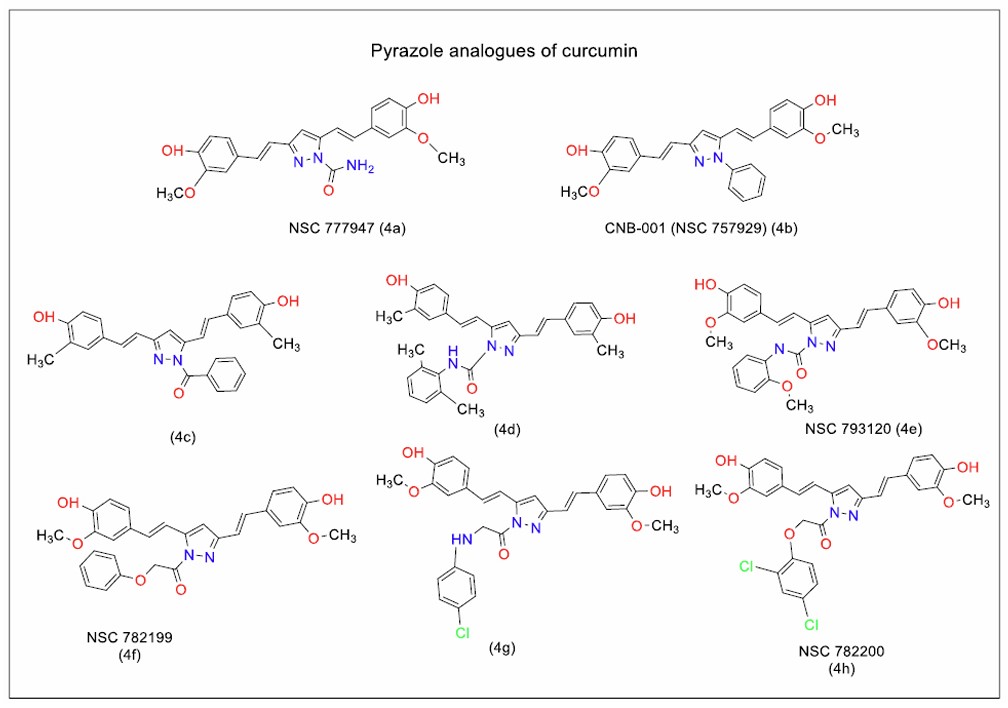
Pyrazole derivatives of curcumin with anticancer activity (Series 4).
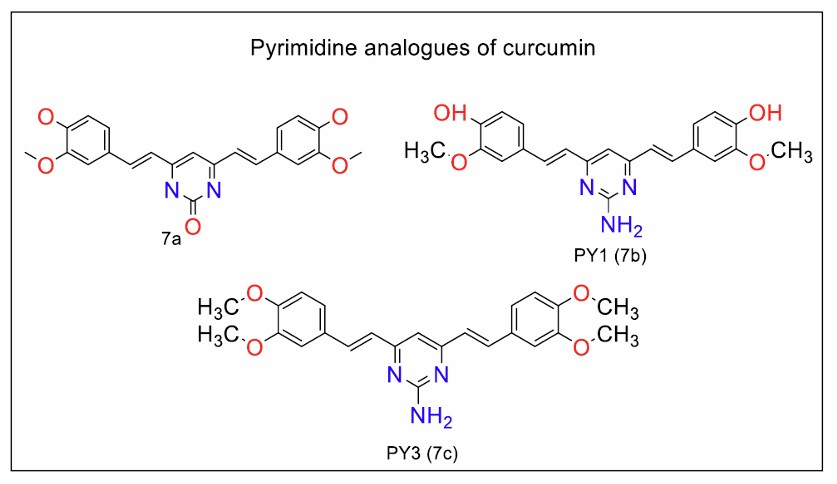
Pyrimidine analogues of curcumin (Series 7).
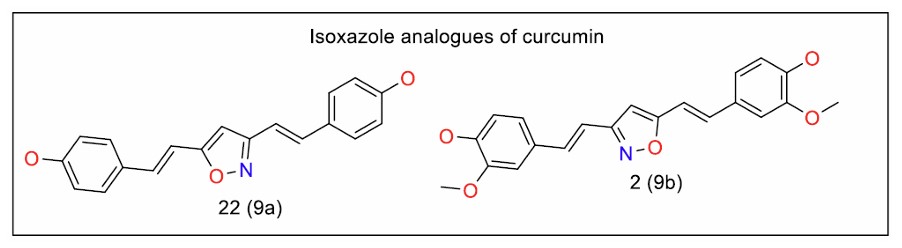
Isoxazole analogues of curcumin (Series 9).
Caprioglio et al. [94] identified curcumin analogues with potent anticancer activity where the diketo group of curcumin was replaced with a triazole ring. The position of the methoxy and phenyl groups on curcumin's phenyl rings was varied to explore the structure-activity relationship. The analogue with the same substitution pattern as curcumin was most potent, suggesting that retaining certain structural elements may be crucial for maintaining and enhancing biological activity.
2.4. Curcumin Analogues with a hepta-1,6-diene-3,5-dione
Yue et al. [95] identified AC17 (Fig. 8, 6c), a 4-arylcurcumin analogue with tenfold more potency than curcumin in inhibiting the growth of multiple cancer cell lines, including nasopharyngeal carcinoma [CNE2] and lung cancer [A549] cells. Ma et al. [96] found that formulating AC17 into transmucosal delivery microneedles coated with hyaluronic acid increased its bioavailability in mice. This formulation successfully blocked the growth of oral squamous carcinoma cells by regulating the FOXO3 signalling pathway.
Hussain et al. [97] identified methyl, methoxy, and chloro-substituted curcumin analogues that exhibited potent antioxidant and antidepressant activities, highlighting the diverse therapeutic potential of curcumin derivatives. Liu et al. [98] elucidated the mechanism by which a curcumin analogue, T63, induces apoptosis and cell cycle arrest in nasopharyngeal cancer cells (CNE2 and CNE2R). The structure of T63 is characterised by an arylidene group at the methylene carbon, which contributes to its efficacy in overcoming cancer cell resistance [99].
Yang et al. [100] studied the potential of ASC-J9 (Fig. 8, 6a), a dimethoxy analogue of curcumin, in treating spinal and bulbar muscular atrophy [SBMA]. The compound regulated the toxicity of the androgen receptor in in vitro, Drosophila, and murine models. Bott et al. [101] identified ASC-JM17 (JM17) (Fig. 8, 6b), an orally bioavailable curcumin analogue with improved efficacy in both in vitro and in vivo models of SBMA. Structural modifications, including reduction of the diketo group to a monoketogroup and substitution of the methylene carbon with a cyclobutane ring, improved the pharmacokinetic profile of this molecule. JM17 also exhibited potent antioxidant activity by regulating Nrf2. Wu et al. [102] compared the efficacy of JM17 (1 µM) to the known Nrf2 activator omaveloxolone (0.3 µM). JM17 was more effective at reducing mitochondrial ROS and lowering glutathione levels.
Seghetti et al. [103] evaluated novel 1,4,6-heptatrien-3-one analogues, incorporating a substituted triazole ring on the active methylene group to pan-assay interference (PAINS) properties characteristic of curcumin. Triazole-substituted analogues on the phenolic hydroxyl group showed less potency than those with triazole substitution on the methylene group. The analogue substituted with a para-fluorobenzyl moiety (Fig. 8, 6d) was the most potent. This compound outperformed curcumin in inducing apoptosis in leukaemia T cell lines, validating the influence of chemical structure on pharmacological activity.
Aloor et al. [104] adopted a computational approach for designing chlorinated curcumin analogues with antiproliferative activity. Docking and molecular modelling tools were used to identify analogues with favourable drug-like properties and stronger binding affinity than curcumin. One analogue showed superior binding interactions with the BRCA1-BRCT-c domain and demonstrated significant anticancer activity against breast cancer cell lines, emphasising its therapeutic potential in breast cancer treatment.
2.5. Dibenzylidenecyclohexaneoxime Analogues of Curcumin
Wang et al. [105] identified JM-2, an oxime analogue of dibenzylidenecyclohexane (Fig. 9, 5a), that demonstrated promising therapeutic effects in murine models of diabetic cardiomyopathy. Its efficacy was linked to NF-κB-mediated inhibition of inflammation, highlighting its potential for treating inflammation-related complications in diabetes.
In another study, Lou et al. [106] discovered SJ-12 (Fig. 9, 5b), a curcumin analogue that may be beneficial in the treatment of cardiomyopathy. The probable mechanism of action is through the regulation of the calcium signalling pathway. In a murine model of streptozocin-induced diabetic cardiomyopathy, SJ-12 did not influence blood glucose levels, suggesting its potential use in cardiac diseases without affecting glucose metabolism.
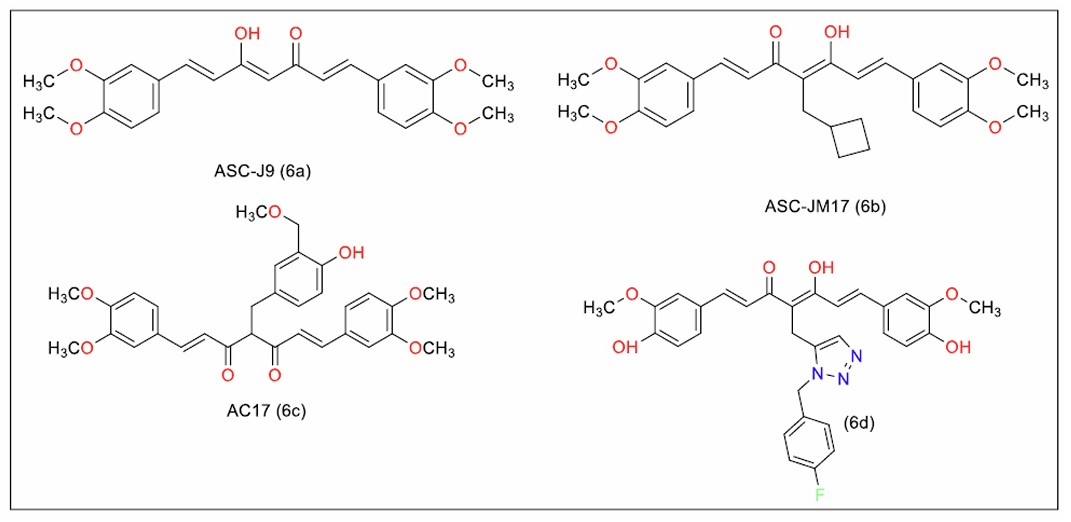
Hepta-1,6-diene-3,5-dione analogues of curcumin (Series 6).
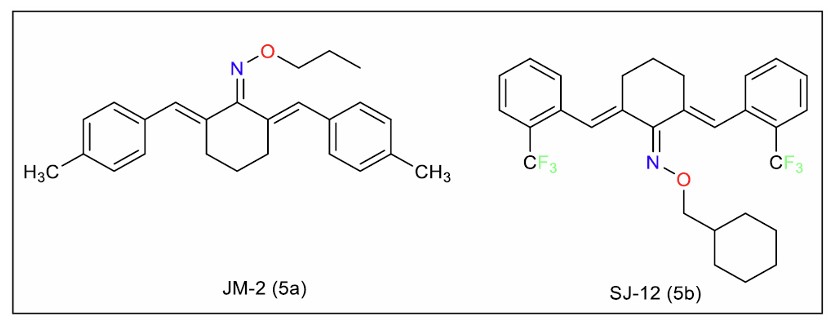
Oxime analogues of curcumin.
2.6. β-cyclocitral Analogue of Curcumin
Han et al. [107] synthesised β-cyclocitral-derived monocarbonyl curcumin analogues. A19 (Fig. 10) exhibited strong cytotoxicity against hepatocellular carcinoma (HCC) cell lines HepG2 and Huh-7. It reduced cell viability, suppressed colony formation, and induced G2/M phase arrest while promoting apoptosis in cancer cells, with high selectivity. A19 induced DNA damage by inhibiting the ERK/JNK/p38 MAPK signalling pathway, which plays a pivotal role in cell survival and stress response.
Combining A19 with sorafenib, a standard chemotherapeutic for HCC, enhanced A19's cytotoxicity, suggesting A19's potential as a combination therapy for liver cancer. A detailed structure-activity relationship [SAR] study further investigated the impact of various substituents on the cyclohexanone ring of A19 analogues. Analogues without substituents exhibited weak activity similar to β-cyclocitral. However, the introduction of halogenophenyl groups, particularly meta-bromophenyl and dihalogenophenyl substituents, improved anticancer activity. Compounds like A8, with para-[tert-butyl] phenyl [IC50 = 35.69 μM], exhibited notable efficacy. Analogue A9 [substituted with 2,5-ditri-fluoromethylphenyl] showed reduced activity. The compound A10, which had an ortho-ethoxy group, exhibited lower potency. These results highlight the effect of electron-withdrawing substituents on activity.
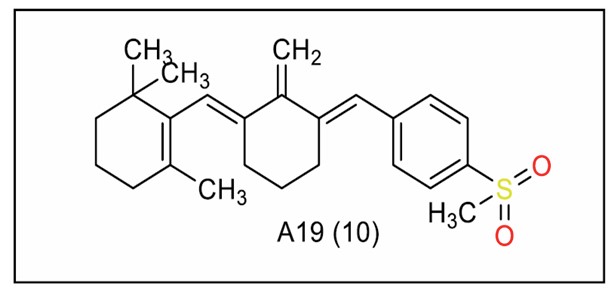
β-cyclocitral-derived mono-carbonyl curcumin analogue (Series 10).
2.7. Azepane Derivatives of Curcumin
Wang et al. [108] synthesised VLX1570 (Fig. 11), an analogue of b-AP15 with increased potency and hydrophilicity. However, the compound faced limitations in clinical trials due to pulmonary toxicity [109]. This was attributed to the presence of the 1,4-diene-3-one moiety, which forms glutathione complexes in vitro [110, 111].
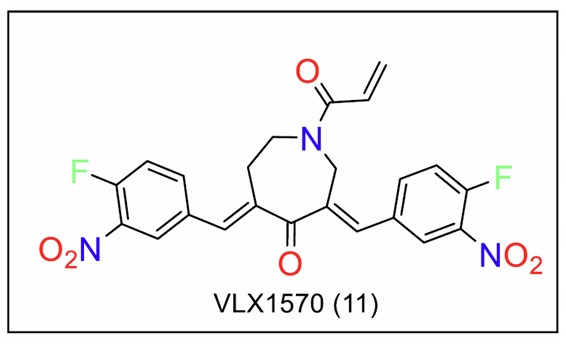
Azepane analogues of curcumin.
2.8. Conjugation Strategies
Various conjugation approaches have been explored to enhance the stability, potency, and solubility of curcumin analogues.
2.8.1. Diflouroborate Conjugates of Curcumin
Kuźmińska et al. [112] investigated the anticancer potential and chemical stability of fluorinated curcumin analogues and their difluoroborate conjugates. Complexing the diketone group of curcumin with difluoroborates and fluorinating the phenyl rings yielded compounds with enhanced potency and stability compared to curcumin. Among the compounds tested, the difluoroborate conjugate with a fluorine atom at the third position and a methoxy group at the fourth position of the phenyl ring showed the most potent anticancer activity.
2.8.2. Amino acid Conjugates of Curcumin
Chan et al. [113] synthesised amino acid conjugates of curcumin to improve its aqueous solubility and assess their proteasome-inhibiting properties. They synthesised conjugates, including bisglycinoyl, bisalaninoyl, and bisvalinoyl, hydrochloride salts of curcumin. The aqueous solutions of these analogues exhibited anticancer activity against the LNCaP prostate cancer cell line. These amino acid conjugates demonstrated proteasome inhibitory activity, further supporting their potential as effective anticancer agents with enhanced solubility.
2.9. Synthetic Routes for Curcumin Analogues
2.9.1. Monocarbonyl Analogues
The general scheme for the synthesis of monocarbonyl analogues of curcumin is represented in Fig. (12). Aromatic aldehydes undergo condensation with cyclic or straight-chain ketones to give the respective analogues.
2.9.2. Synthesis of Heterocyclic Analogues of Curcumin
Figure 13 illustrates the synthesis of curcumin analogues where the central diketo group of curcumin is replaced with various heterocyclic rings, such as pyrazoles, isoxazoles, pyrimidines, and triazoles. These transformations generally involve cyclisation reactions facilitated by reagents such as hydrazines, hydroxylamine, or azides, under reflux or buffered conditions.
2.9.3. General Scheme for the Synthesis of hepta-1,6-diene-3,5-dione Analogues
Figure 14 represents the boron-mediated aldol condensation and subsequent functional group modification protocol used to synthesise hepta-1,6-diene-3,5-dione analogues of curcumin. The process begins with the formation of a boron complex from acetylacetone and boric acid, facilitating the condensation with substituted aromatic aldehydes in the presence of base and acid to construct the conjugated dione scaffold characteristic of curcumin analogues. Further, this intermediate can undergo modifications such as piperidine/AcOH–catalysed condensation with additional aldehydes for structural extension, catalytic hydrogenation using palladium on carbon to saturate carbon–carbon double bonds, and reduction with lithium aluminium hydride (LiAlH4) in THF to yield hydroxy derivatives. Through these steps, a diverse array of curcumin analogues with altered core motifs and functionalities is produced, aiming to enhance metabolic stability and modulate biological activity compared to native curcumin [95].
2.9.4. Synthesis of the Azepane Derivative of Curcumin
The synthesis of VLX 1570 (Fig. 15) involves a multi-step procedure beginning with the formation of a key intermediate via condensation of 4-perhydroazepinone hydrochloride with 4-fluoro-3-nitrobenzaldehyde under acidic conditions for around 100 hours. This intermediate is then functionalized by coupling with alkenyl carboxylic acids using propylphosphonic anhydride as a coupling reagent, typically in the presence of triethylamine and tetrahydrofuran solvent at low temperature.
3. COMPUTATIONAL ADME PROFILING AND DOCKING STUDIES OF THE CURCUMIN ANALOGUES
Curcumin analogues are a diverse set of compounds whose experimental activity data are inconsistent across various studies. This variability across activity makes computational prediction tools valuable for generating standardised in silico data for guiding further development [114]. We used computational approaches to compare the performance of curcumin analogues, recognising that in silico predictions can highlight trends even where experimental measurements are lacking or not directly comparable. Curcumin was included as a reference compound. Predicted ADME properties [absorption, permeability, P-gp substrate, and key CYP450 interactions] were evaluated to assess drug-likeness.
Our analysis focused on the kinase DYRK2, a serine/threonine kinase that has been co-crystallised with curcumin. DYRK2 is a biologically relevant target. Curcumin binds to DYRK2 and potently inhibits its activity [with curcumin showing low-nanomolar DYRK2 inhibition], leading to downstream effects on cancer proteasome function and proliferation (5). DYRK2 has been implicated in multiple cancers and neurological disorders [115], underlining its therapeutic significance.
DYRK2 regulates the function of the 26S proteasome by phosphorylating RPT3, an essential component of protein degradation. Inhibition of DYRK2 impairs proteasome activity, leading to protein accumulation and cell death. DYRK2 also phosphorylates heat shock factor 1 [HSF1]. This promotes the expression of chaperone proteins to manage proteotoxic stress, especially in triple-negative breast cancer [116].
DYRK2 regulates apoptosis by facilitating the ubiquitination of MOAP-1, which activates the pro-apoptotic protein Bax. This contributes to programmed cell death and cellular homeostasis. Curcumin directly inhibits DYRK2 by binding its ATP-binding pocket, as confirmed by high-resolution crystallography [116]. This indicates the potential of curcumin and its analogues as anticancer agents.
DYRK2’s role in cancer involves maintaining proteostasis, regulating apoptotic pathways, and modulating stress responses. This makes it a vital target in the treatment of various cancers, including breast and TNBC [116].
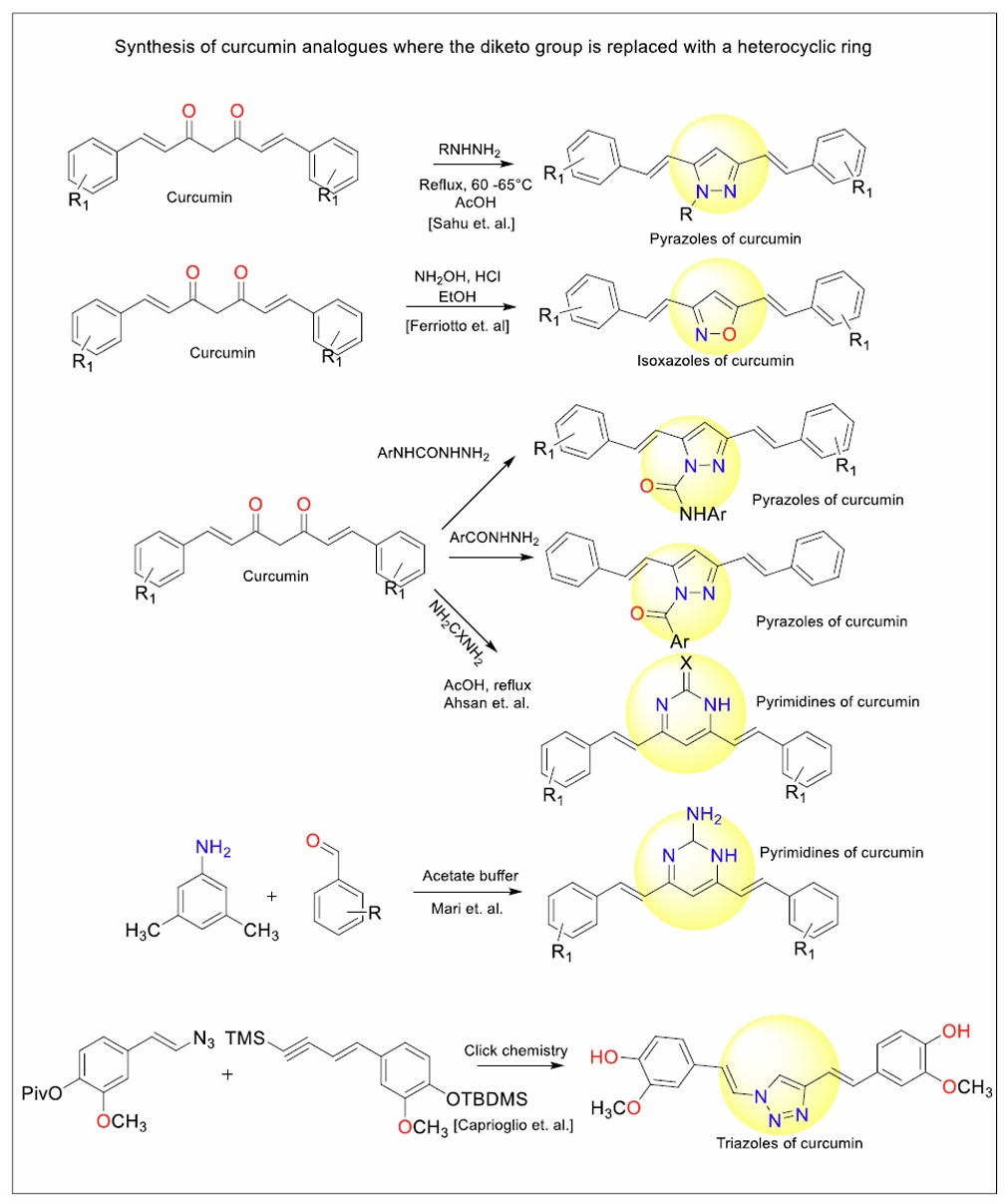
General scheme for the synthesis of heterocyclic analogues of curcumin.
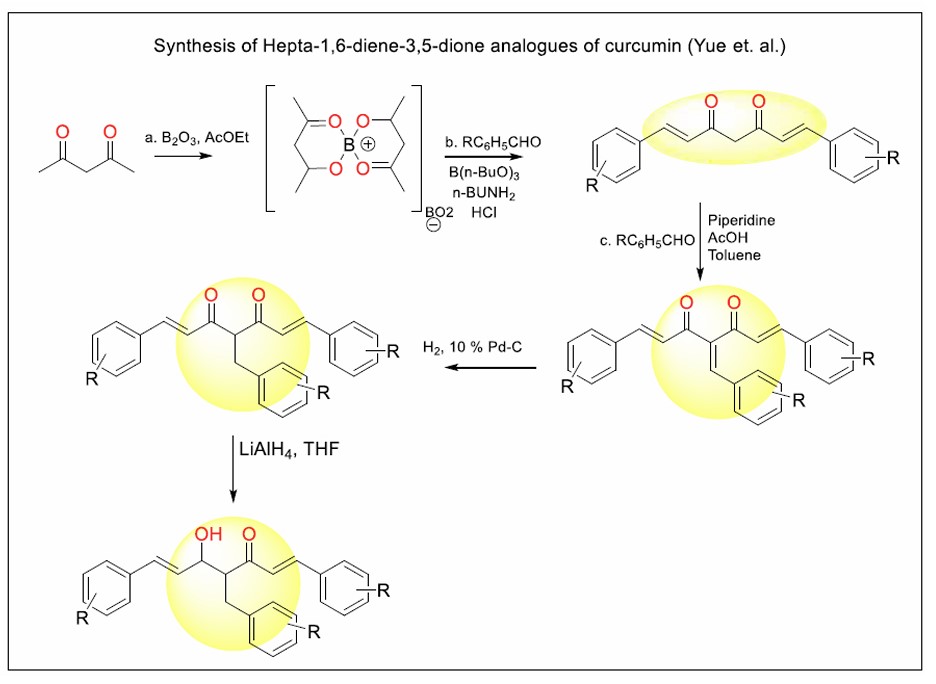
General scheme for the synthesis of hepta-1,6-diene-3,5-dione analogues of curcumin [95].
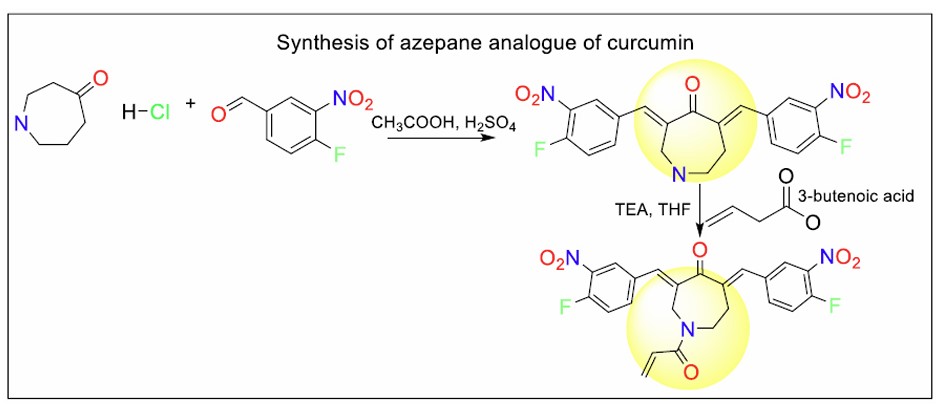
Scheme for the synthesis of the azepane analogue of curcumin.
The availability of a curcumin-bound DYRK2 crystal structure provides a rational basis for docking these analogues and for observing key binding-site interactions [e.g., within the ATP pocket]. Here, we discuss the structure–activity relationships [SAR] of curcumin analogues docked in DYRK2, clustered by their chemical scaffolds. The predicted binding metrics and ADME properties are analysed together to identify the most promising candidates [116].
3.1. Methodology
LigPrep was used to prepare the compounds and ensure proper geometry and protonation states. Neutral forms of molecules were used for analysis to align with typical physiological conditions. QikProp was used to predict the properties in batch mode.
QikProp calculates various molecular descriptors, including molecular weight, hydrogen bond acceptor/donor counts, octanol/water partition coefficient [QPlogPo/w], aqueous solubility [QPlogS], Caco-2 cell permeability [QPPCaco], blood-brain barrier penetration [QPlogBB], human oral absorption [PHOA], and Lipinski's Rule of Five violations, among others. The output data was exported into Excel for further analysis of ADME properties [117].
Molecular docking of curcumin analogues was performed using the Maestro suite (118). Protein preparation of the DYRK2 protein [PDB ID: 6HDR] was done at a simulated pH of 7.4. Protein preparation involved filling missing side chains, optimising hydrogen bond assignments, and minimising/deleting water molecules. The Receptor Grid Generation module in GLIDE was used to generate a grid around the ligand-binding site.
The docking protocol was validated by redocking the co-crystallised ligand into the prepared grid using both Standard Precision [SP] and Extra Precision [XP] docking modes. The docked poses were superimposed onto the original structure of the co-crystallised ligand to identify the most suitable docking mode. The XP docking pose showed the minimum root mean square deviation [RMSD] value of 0.363 Å compared to the crystallographic pose (Fig. 16), indicating a valid docking setup.
The data from in silico analysis is available as a supplementary file.
The curcumin analogues, prepared with LigPrep, were docked flexibly using XP docking into the DYRK2 receptor. Only the top-scoring pose per ligand was retained. Per-residue interaction scores for amino acid residues within 4 Å of the grid centre were recorded and analysed to understand key ligand-protein interactions.
The SMILES representations of the curcumin analogues were input into the SWISS ADME web tool to predict their cytochrome P450 [CYP] enzyme interactions and to identify potential substrates of P-glycoprotein [P-gp] [119].
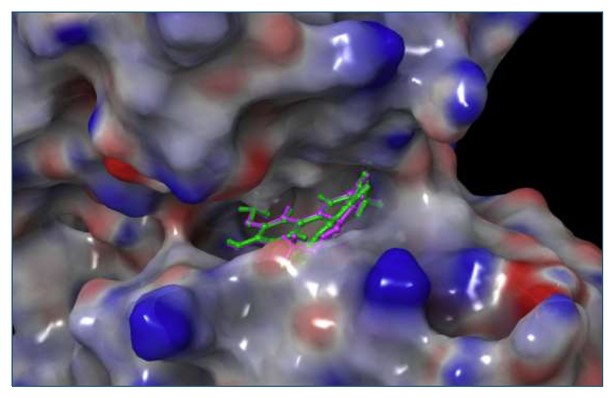
Docking validation against DYRK2 protein.
3.1.1. Piperidone Analogues
Piperidone analogues [1a–1m] feature a six-membered piperidone ring linking two aromatic moieties. This class showed generally strong predicted binding to DYRK2. Most Piperidones outperformed curcumin [XP GScore -5.1]; the best, 1j, achieved an XP GScore of -10.46 with high ligand efficiency. Analogue 1j [bearing N, N-dimethylamino and alkoxy substituents] maximises hydrophobic contacts in the ATP pocket – notably engaging Ile294 deeply and contacting Val163 and Ala176 on the kinase hinge. Other analogues, such as 1g and 1k, also scored well [~ -9.0] due to bulky hydrophobic groups accessing residues like Ile155 and Ile294. In contrast, highly polar substitutions [e.g., in 1h and 1i] led to much weaker binding [~ -4 to -5], indicating that excessive polarity or flexibility disrupts interactions in the predominantly hydrophobic site.
From an ADME perspective, piperidones have high predicted oral absorption [>80%], and none are P-gp substrates. However, nearly all are predicted to inhibit CYP2C19 and CYP2C9, and the analogues 1a–1d may also inhibit CYP1A2. Thus, despite excellent DYRK2 affinity, the piperidone scaffold exhibits broad CYP liabilities that may require structural tuning.
3.1.2. Aliphatic Analogues
The aliphatic series [2a–2j], with flexible acyclic linkers resembling curcumin’s heptadienone chain, showed a wide range of docking results. Several analogues achieved excellent scores, for example, 2d and 2f [XP GScores ≈ -10.4], indicating that suitable substituents or conformational constraints can significantly enhance binding even on a flexible scaffold. 2j also scored well [~ -9.2], reinforcing that an aliphatic linker can align optimally given proper orientation. By contrast, analogue 2e was a clear outlier, with a significantly weaker score [-6.1], and was uniquely predicted as a P-gp substrate. The poor performance of 2e suggests that its structure [perhaps overly polar or flexible] fails to engage the pocket effectively and is prone to efflux.
ADME-wise, aliphatic analogues generally have high permeability and absorption [~100%]. Apart from 2e, none are P-gp substrates. Metabolically, however, this class tends to incur multiple CYP inhibitions: most analogues are flagged for CYP2C19 and CYP2C9 inhibition, and some [e.g., 2a, 2d] may also inhibit CYP1A2 or CYP3A4. Thus, while several aliphatic analogues rival the potency of rigid scaffolds, their high lipophilicity corresponds with broad CYP interference that would need to be addressed.
3.1.3. Cyclopentanone Analogues
Cyclopentanone analogues [3a–3h], which incorporate a rigid five-membered ketone linker, were among the strongest binders. This cyclic scaffold pre-organises the molecule for optimal binding. Multiple cyclopentanones achieved very high scores [e.g., 3d -10.9; 3a, 3g ~ -10.7 to -10.8], far better than curcumin. Locking the linker in a planar conformation appears to maximise hydrophobic contacts with key residues [Ile155, Val163, Leu282, Ile294]. Only 3e and 3h showed substantially lower affinity [~ -5.8], suggesting that certain substituents in those two [possibly adding flexibility or polarity] disrupt the favourable binding geometry of the cyclopentanone core.
ADME predictions for cyclopentanones are largely positive: high absorption [>85%] and no P-gp efflux. Like other hydrophobic analogues, however, this class shows a tendency for CYP inhibition – nearly all 3-series compounds are flagged for CYP2C19 and 2C9, and several [especially 3b–3h] for CYP2D6 – though none are predicted to inhibit CYP3A4. In summary, the cyclopentanone scaffold yields some of the most potent DYRK2 binders; however, further optimisation would be needed to mitigate their broad CYP liabilities.
3.1.4. Pyrazole Analogues
Pyrazole analogues [4a–4h] use a planar five-membered heterocycle in place of curcumin’s diketone. This generally preserved strong binding: most pyrazoles docked with high affinity [average scores around -8 to -9]. In the case of compound 4a [XP GScore -10.2, high efficiency], a pyrazole linker has optimised the pharmacophore for tight binding. The docking score of 4c was weakest [-5.4], and 4d was intermediate [-6.0]. Compound 4d is predicted not to inhibit any major CYP and is not a P-gp substrate [a clean ADME profile]; however, it has moderate potency. This contrast within the pyrazole series demonstrates that small substituent differences can have a dramatic influence on pharmacokinetic properties.
Pyrazole analogues are well absorbed and avoid P-gp-mediated efflux. Several of the more potent examples [e.g., 4b, 4f, 4g] are predicted to be for CYP2C19 and CYP2C9 inhibitors. None of the pyrazole analogues is associated with CYP1A2 or CYP3A4 issues. The relatively moderate profile of 4d [aside from 2C9] suggests the pyrazole template can be explored to achieve a balance between potency and drug-likeness by careful substituent selection.
3.1.5. Cyclohexanone Analogues
Cyclohexanone analogues [5a–5e] have been predicted to be potent DYRK2 binders. The rigid six-membered linker optimises hydrophobic contacts, giving them the highest docking scores among the other analogues. 5c had an XP GScore of -11.53 [the best overall] with the highest binding efficiency. 5b and 5d were nearly as potent [~ -11.2 and -10.8], indicating the cyclohexanone core can fully exploit the ATP-site pocket. However, 5e was a notable exception with a much weaker score [~ -5.0], implying that its substituents or geometry prevented optimal binding.
ADME profiles for cyclohexanones resemble those of cyclopentanones. The predicted absorption is high [~90–100%], and none of the analogues are P-gp substrates. All analoguesare predicted to be inhibitors of CYP2C19 and CYP2C9. None are predicted to inhibit CYP2D6 or CYP3A4. In summary, the cyclohexanone scaffold stands out for binding potency, although its universal CYP2C inhibition needs to be addressed. The anomalous drop in the binding score of 5e underscores the importance of appropriate substituents for optimum binding affinity.
3.1.6. Heptadienone Analogues
Heptadienone analogues [6a–6d] retain curcumin’s flexible 7-carbon linker. Compound 6a was quite potent [XP GScore ~ -9.8, much better than curcumin’s -5.1], whereas 6c and 6d were much weaker [~ -5 to -6]. Notably, 6c had the lowest score in this class and was among the few analogues predicted to be P-gp substrates. The highly polar or bulky nature of the substituents could contribute to weak binding and permeability.
The heptadienones show good absorption [~90%+] and, aside from 6c, no P-gp issues. Analogues 6a and 6b are flagged as CYP3A4 inhibitors [a liability not seen in the rigid scaffolds], in addition to the common CYP2C9 inhibition. Compounds 6c and 6d are also predicted to inhibit CYP2C19. The high affinity of these analogues often coincides with broader CYP inhibition (6a) and, in the case of 6c, with efflux, underscoring the challenges posed by this linker.
3.1.7. Pyrimidine Analogues
Replacement of the linker with a pyrimidine ring [7a–7c] proved highly effective. All three analogues docked strongly [XP GScores ~ -8 to -10.7]. Compound 7a was best [~ -10.7], and even the weakest binder, 7c [~ -7.9], outperformed curcumin. The planar pyrimidine core likely positions the aryl rings favourably, and its ring nitrogens may enable a classic kinase hinge interaction to enhance binding.
ADME predictions indicate that the pyrimidines have good absorption [≈approximately 80–90%] and no P-gp efflux. Their CYP profile is somewhat cleaner than most classes: like others, they inhibit CYP2C9, but only 7c [the least potent] is flagged for inhibition of CYP2C19 and CYP3A4. 7a and 7b avoid major CYP issues beyond 2C9. Thus, the pyrimidine scaffold emerges as a promising motif combining strong target affinity with a slightly more selective ADME profile in its top analogues.
3.1.8. Oxime Analogues
Oxime analogues [8a, 8b] convert curcumin’s diketone to an oxime. This change had opposite effects on binding: 8a docked moderately well [XP GScore ~ -7.7], whereas 8b was extremely weak [-2.7, the worst of all]. The stark contrast indicates that multiple polar oxime moieties [as in 8b] can disrupt planarity and binding, whereas a single oxime [8a] is tolerated.
Both oximes are predicted to have high absorption [>90%], and neither is a substrate for P-gp. However, both are flagged as inhibitors of CYP2C19, 2C9, and 3A4. Given their broad CYP liabilities and limited binding improvement, the oxime modification appears less attractive for this series.
3.1.9. Isoxazole Analogues
Isoxazole analogues [9a, 9b] – featuring a five-membered isoxazole ring as the linker – performed exceptionally well. Both 9a and 9b achieved XP GScores around -10 or better; 9b reached -10.8 [among the top overall]. The planar isoxazole [with one N and one O atom] likely positions the molecule optimally without sacrificing hydrophobic surface area, resulting in a very tight fit in the pocket.
ADME-wise, 9a and 9b are predicted to be well absorbed and non-P-gp. They also show a cleaner CYP profile than most analogues. Apart from the ubiquitous CYP2C9 inhibition, the isoxazoles have few liabilities: 9a is flagged for CYP1A2, while 9b is not predicted to inhibit any CYPs beyond 2C9 to a significant extent. With its combination of excellent binding and limited metabolism alerts, 9b stands out as one of the most promising candidates in the entire series.
3.1.10. Citral Analogue
Citral-derived analogue [10] showed only a moderate docking [XP GScore -5.8, similar to curcumin]. The low binding affinity could be due to its flexible aliphatic nature. Its ADME profile is favourable, with 100% absorption and no P-gp efflux. However, it is predicted to inhibit CYP2C9 and 3A4. Thus, the citral-based design did not improve potency or reduce metabolic risk compared to curcumin.
3.1.11. Azepine Analogue
The Azepine analogue [11], containing a seven-membered heterocycle linker, was one of the weakest binders [XP GScore -3.2]. The bulky ring may cause excessive flexibility or steric hindrance, resulting in poor binding. The compound is predicted to be well absorbed with no P-gp issues. However, it is predicted to inhibit CYP2C9 and CYP3A4. Given its low efficacy and metabolic liabilities, the azepine modification appears unfavourable for further development.
4. CURCUMIN–GLUTATHIONE INTERACTIONS: ELECTROPHILIC REACTIVITY AND IMPLICATIONS FOR DRUG DESIGN
Interaction with glutathione is one factor that influences a compound's activity profile. Reactions with glutathione occur both spontaneously and via conjugation by glutathione S-transferase [GST] enzymes. This conjugation facilitates detoxification of reactive curcumin metabolites. Curcumin influences cellular redox homeostasis by inducing the expression of GST and enzymes involved in GSH biosynthesis. This upregulation helps maintain intracellular GSH levels, balancing its levels during conjugation and protecting against oxidative stress [120].
Electrophilic reactivity with GSH is probably at the root of curcumin’s promiscuity for covalent protein binding. Structural modifications that reduce electrophilicity may mitigate adverse effects without compromising therapeutic efficacy. One promising approach is to introduce heterocycles between the benzylidene moieties of curcumin analogues to enhance metabolic and chemical stability. However, the dienone moiety remains a key factor in the promiscuity of monoketone curcumin analogues, as established by Ward et al [111].
Predictions using XenoSite (a tool for assessing chemical reactivity) confirm that curcumin and the azepine analogue [11] demonstrate significant reactivity with glutathione, cysteine residues in proteins, and DNA interactions [111]. Exploring the reactivity of other curcumin analogues through XenoSite also revealed that many reported analogues retain this reactivity, underscoring the need for further structural modifications to reduce off-target effects.
5. FUTURE DIRECTIONS
The advantage of curcumin and its analogues is the extensive literature on their synthesis, bioactivity, bioavailability, and potential toxicity. This information provides a strong foundation for further optimisation of these compounds. However, there remain critical areas for improvement.
Rigid and aromatic linkers [piperidone, cyclopentane/ hexanone, pyrazole, pyrimidine, isoxazole] generally conferred higher DYRK2 affinity than flexible or highly polar modifications. Several analogues from these classes [notably 5c, 9b, 7a, 3d, 1j] have very favorable docking scores, maintaining crucial hydrophobic contacts [e.g., with Ile155, Val163, Leu282, Ile294]. However, many high-affinity analogues also inhibit multiple CYP enzymes [especially 2C9 and 2C19]. This indicates a trade-off between potency and metabolic selectivity. One compound from the isoxazole analogues, 9b, stands out for combining high binding affinity with a relatively low number of CYP interactions, making it a strong lead for further development. An analogue like 4d, while less potent, demonstrates that minimizing aromatic bulk and polar groups can yield a very clean ADME profile. These insights provide a roadmap for further structural modifications of curcumin. A combination of structural features that optimise DYRK2 binding and reduce ADME liabilities can yield curcumin analogues with translational potential.
Modification of the diene or keto groups in curcumin analogues is one strategy to reduce their promiscuity. The likelihood of reactions with glutathione or proteins can be addressed through targeted molecular adjustments. Although replacing the diketo group of curcumin with aromatic heterocycles does not significantly decrease reactivity with glutathione or protein adduct formation, these analogues may still hold value for alternative therapeutic applications. These analogues could be explored as candidates for topical treatments in conditions such as skin infections or eczema, where localised activity may be required. These insights open the door to designing curcumin analogues with lower off-target reactivity while optimising their therapeutic potential.
Boron complexes of curcumin have been synthesised with the diketone group of curcumin complexed with boron. The stability and solubility of these complexes at different pH ranges need to be evaluated.
CONCLUSION
Despite the widely studied pharmacological activity of curcumin in a wide range of diseases, its poor pharmacokinetics and promiscuity limit its clinical applications. Low bioavailability and rapid metabolism have hindered its development. Various structural modifications have been explored to improve these limitations, including the development of mono-ketone analogues, monoketene analogues, and heterocyclic derivatives. These modifications have largely aimed at enhancing the chemical and metabolic stability, solubility, and bioactivity of curcumin analogues. Some of these analogues have even advanced to in vivo studies and clinical trials.
Curcumin analogues, such as those incorporating heterocycles like imidazole, triazole, or pyrrole between the benzylidene moieties, have demonstrated improved stability and solubility compared to curcumin. The difluoroborate conjugates and amino acid conjugates have shown significant improvements in aqueous solubility and anticancer activity. While these advances mark a step forward, issues surrounding the toxicity, selectivity, and efficacy of curcumin analogues remain unresolved. Several analogues exhibit undesirable reactivity, especially toward glutathione, cysteine residues in proteins, and DNA, due to the dienone moiety, which limits their clinical applicability.
Several critical research gaps need to be addressed to further advance curcumin analogues. The areas for improvement include therapeutic selectivity and target-specific efficacy. Efforts should focus on refining molecular interactions through structure-based drug design and docking studies, particularly for targets such as DYRK-2 and other ubiquitin-specific proteases. Long-term toxicity profiling and more rigorous in vivo studies are needed to evaluate the safety and off-target effects of these compounds at therapeutic doses.
While a few curcumin analogues have faltered in clinical trials, many are still in the preclinical phase. Significant optimisation is required to improve bioavailability, selectivity, and therapeutic index. Novel chemical modifications could offer solutions to these challenges. By addressing these gaps, the therapeutic potential of curcumin and its analogues can be fully realised, thereby improving hit-to-lead generation.
Despite the progress made, the path to translating curcumin and its analogues into clinical applications remains challenging. The existing body of literature provides a strong foundation for designing more effective and safer analogues. However, there is still much work to be done to overcome the limitations in efficacy, stability, and selectivity. Continued research and structure optimisation could lead to the identification of efficient and selective curcumin analogues that progress into clinical trials.
AUTHORS’ CONTRIBUTIONS
The authors confirm contribution to the paper as follows: A.U.W.: Conceptualisation, Methodology, Writing-Original draft Visualisation; K.K.M.: Data Curation, Methodology, Writing – Review & Editing; M.K.B.: Data Curation, Methodology, Writing – Review & Editing; S.Z.: Writing - reviewing and editing; J.C.P.: Methodology, Visualisation; M.R.P.: Supervision, Writing – reviewing and editing. All the authors reviewed and approved the final version of the manuscript.
ABBREVIATION
| ROS | = Reactive Oxygen Species |
ACKNOWLEDGEMENTS
Declared none.

