All published articles of this journal are available on ScienceDirect.

Covalent Inhibition of Thioredoxin Reductase by Michael Acceptors: Rational Design, Synthesis, and Biological Evaluation of Oxazol-5(4H)-one Derivatives
Authors Info & Affiliations
Abstract
Introduction
Thioredoxin reductase 1 (TrxR1) is a key enzyme involved in maintaining redox homeostasis in cancer cells and is considered a promising target for anticancer drug development. This study aimed to rationally design, synthesize, and evaluate a novel series of oxazol-5(4H)-one derivatives as potent and selective inhibitors of TrxR1.
Methods
The design process was initiated by screening a commercial library of oxazolones, which informed the construction of a QSAR model. Guided by molecular docking studies against the TrxR1 catalytic site (PDB: 3EAN), a focused library of 18 target 4-benzylidene-oxazol-5(4H)-ones was synthesized via a multicomponent reaction using substituted hippuric acids and aromatic aldehydes. The inhibitory activity against TrxR1 and the off-target enzyme glutathione reductase (GR) was determined using a DTNB assay on A549 cell lysates. Cytotoxicity was evaluated against a panel of human cancer cell lines (A549, SHSY5Y, U251, and HELA) and normal HEK293T cells using an MTT assay. The mechanism of action was further investigated through immunoblotting analysis.
Results
Several compounds demonstrated potent and selective TrxR1 inhibition. Lead compounds 1i, 1o, and 1s exhibited significant TrxR1 inhibition (over 50% at 25 µM) with low activity against GR. The IC50 values for TrxR1 inhibition were in the nanomolar range (0.25 nM for 1i, 19.7 nM for 1o, and 4.2 nM for 1s). These compounds also showed pronounced cytotoxicity against cancer cells, with compound 1i being the most active against U251 glioblastoma cells (IC50 = 12.58 µM) and demonstrating selectivity over normal cells. Immunoblotting analysis confirmed that inhibitors containing dual Michael acceptor motifs (e.g., 1q-s) induced the formation of covalent TrxR1 dimers, indicative of a unique mechanism of action.
Discussion
The results validate our rational design strategy, highlighting the critical role of the Michael acceptor warhead and the oxazolone core in effective TrxR1 binding and inhibition. The high selectivity for TrxR1 over GR and the observed cytotoxic selectivity toward cancer cells underscore the therapeutic potential of this compound class. The discovery of TrxR1 dimerization provides a novel mechanism for irreversible enzyme inhibition.
Conclusion
A new series of oxazol-5(4H)-one derivatives was successfully developed as highly potent and selective TrxR1 inhibitors. The lead compounds 1i, 1o, and 1s showed promising anticancer activity in vitro, coupled with a favorable selectivity profile. This work not only establishes a robust structure-activity relationship but also introduces a rational strategy for designing novel TrxR1-targeted antitumor agents, supported by the discovery of a dimerization-based inhibition mechanism.
1. INTRODUCTION
The thioredoxin system-comprising thioredoxin (Trx), thioredoxin reductase (TrxR1), and NADPH-is a central redox regulatory network essential for maintaining cellular homeostasis in mammals [1]. TrxR, a selenocysteine-containing flavoenzyme, catalyzes the reduction of oxidized thioredoxin, thereby influencing critical processes such as DNA synthesis, transcription factor regulation, and antioxidant defense. In cancer cells, the thioredoxin system is frequently overexpressed, supporting tumor survival, proliferation, and resistance to therapy [2]. TrxR, in particular, has emerged as a compelling molecular target for anticancer drug development due to its pivotal role in mitigating oxidative stress and its elevated activity in many malignancies [3]. Elevated TrxR expression is associated with poor prognosis in cancers such as breast, hepatocellular, and lung carcinomas, where it contributes to enhanced DNA synthesis, inhibition of apoptosis, and promotion of angiogenesis and metastasis. The enzyme’s unique C-terminal redox-active motif, containing a reactive selenocysteine residue (Sec498 in human TrxR1), provides a selective site for pharmacological intervention. Inhibition of TrxR disrupts cellular redox balance, leading to accumulation of reactive oxygen species (ROS), oxidative damage, and ultimately cancer cell death [4]. Historically, TrxR inhibitors have been dominated by metal-based compounds and covalent modifiers. Gold complexes such as auranofin, platinum agents like cisplatin, and organoselenium compounds such as ethaselen (BBSKE) have demonstrated potent TrxR inhibition and anticancer activity [5]. However, these agents often suffer from limitations including off-target effects, systemic toxicity, and irreversible enzyme inactivation, which may hinder their clinical utility. Covalent inhibitors that target the Sec residue also risk cross-reactivity with cysteine residues in other proteins, compromising selectivity [6]. Examples of structures of known thioredoxin reductase inhibitors are shown in Fig. (1). This introduction highlights the biological and therapeutic significance of TrxR1, reviews the classes and mechanisms of known inhibitors, and emphasizes the emerging promise of non-covalent inhibition strategies. As our understanding of TrxR1 biology and inhibitor design evolves, continued innovation in medicinal chemistry will be crucial to translate these insights into safe and effective therapies for cancer.
Structure of known michael acceptors covalent TrxR1 inhibitors.
Michael acceptors have significant potential as inhibitors of thioredoxin reductase [3, 7, 8]. TrxR1 is an important target for the development of new anticancer drugs [9-12]. This enzyme plays a critical role in suppressing oxidative stress in neoplastic tissue. A highly selective, drug-like inhibitor of TrxR1 is of considerable importance in medicinal chemistry for the development of new cytotoxic molecules that act through an apoptosis-promoting mechanism.
In one of our chemical programs focused on the synthesis of diazo compounds, we obtained interesting spirocyclic compounds containing a butenolide fragment, which showed high inhibitory activity against thioredoxin reductase [13]. Our search for new Michael acceptors capable of acting as thioredoxin reductase inhibitors was subsequently continued, and the latest results regarding the design, synthesis, and biological activity of the obtained oxazolones are presented in this manuscript.
Oxazol-5(4H)-one is a very important scaffold for diversity-oriented organic synthesis (DOOS) and rational drug discovery. [14, 15]. Oxazol-5(4H)-ones have a different patterns of biological activity [16-18]. These substances play a key role in the synthesis of many small molecules, including amino alcohols, amino acids, thiamine, amides, peptides, and dyes [19, 20]. Many oxazol-5(4H)-ones have important biological activities: antimicrobial [20], anticancer [20], immunomodulatory [21], anti-HIV [22], antiangiogenic [22], antidiabetic [23], insecticidal [24]. Some oxazol-5(4H)-ones act as inhibitors of NO synthase [25], tyrosinase [26], and cyclooxygenase [27]. Oxazol-5(4H)-ones are attractive scaffolds in terms of their compactness and potential for synthetic diversity. [28]. Synthetic approaches of combinatorial chemistry can be successfully applied to the synthesis of oxazol-5(4H)-ones [29]. The possibility of obtaining oxazol-5(4H)-ones with an electron-deficient olefin (so-called Michael acceptor) very important for our research work [30]. From the perspective of medicinal chemistry and early-stage drug development, oxazolones are of particular interest due to a number of key advantages: High degree of structural tenability, the ability to vary substituents at peripheral positions allows fine-tuning of the electronic and steric properties of the ylidene fragment, which directly influences the selectivity and activity of target inhibitors. Due to the accessibility of starting materials and synthetic simplicity, oxazolones can be derived from commercially available or easily synthesized precursors, enabling efficient and scalable construction of chemical libraries. Compliance with Lipinski's rule of five at the design stage, compounds within this class can be pre-designed to adhere to the main parameters of drug-likeness (Lipinski’s rule of five), thereby increasing the likelihood of success in subsequent preclinical studies. Thus, oxazolones represent a promising molecular scaffold for the targeted synthesis and optimization of biologically active compounds in the early stages of developing new therapeutic agents.
2. METHOD
2.1. General Experimental Procedure
All commercial reagents were used without purification. Oxazol-5(4H)-ones were obtained from ChemDiv Corporation and supported with NMR and analytical data. NMR spectra for synthetic compounds were recorded using a Bruker Avance III spectrometer in CDCl3 (1H: 400 MHz; 13C: 100 MHz; 19F: 376 MHz); chemical shifts are reported in parts per million (δ, ppm). The residual solvent peak (CHCl3 or DMSO-d6) was used as internal standard: 7.26 and 2.50 ppm for 1H in CDCl3 and DMSO-d6, respectively; 77.16 and 39.52 ppm for 13C in CDCl3 and DMSO-d6, respectively. Multiplicities are abbreviated as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad, dd = doublet of doublets, dt = doublet of triplets, ddd = doublet of doublets of doublets; coupling constants (J) are reported in Hz.
Mass spectra were recorded using a Bruker microTOF spectrometer (ionization by electrospray, positive ion detection). Melting points were determined in open capillary tubes using a Stuart SMP50 automatic melting point apparatus. Analytical thin-layer chromatography was carried out on UV-254 silica gel plates using appropriate eluents. Compounds were visualized under UV light at 254 nm. Column chromatography was performed using silica gel Merck grade 60 (0.040-0.063 mm; 230-400 mesh) with gradient elution using n-hexane-acetone from 20:1 to 1:2.
TrxR activity measurement was carried out using a DTNB assay; the initial reaction rate was defined as the increase in optical density per minute. Glutathione reductase (GR) activity was determined using a GR activity assay kit (Abcam). Optical density was measured using a CLARIOstar Plus plate reader spectrophotometer (BMG Labtech, Germany). Cell lines A549 (ATCC CCL-185™) and HEK293T (ATCC CRL-3216™) were obtained from the IKBFU cell library.
2.2. General Procedure for Preparation oxazol-5(4H)-ones
Corresponding hippuric acid (1 mmol) was placed in a 25 mL round-bottom flask equipped with a magnetic stirrer. Then, 1 mmol of aromatic aldehyde, 2.5 mmol of anhydrous sodium acetate, and 2.5 mmol of propionic anhydride were added dropwise. The reaction mixture was heated under an inert atmosphere for 4 hours and poured into cold 40% ethanol. A solid formed, and the reaction mixture was filtered off. The product was harvested from the filter and dried at room temperature in a vacuum desiccator.
2.3. NMR and HRMS Spectral Data for Compounds 1g-s
2.3.1. (Z)-4-(4-methoxybenzylidene)-2-phenyloxazol -5(4H)-one 1g
Following GP1 using benzoylglycine (179 mg, 1 mmol) and 4-methoxybenzaldehyde (136 mg, 1 mmol) gave 1g as a yellow solid; yield: 97 mg (35%); m.p. 158.1-159.4 °C.
1H NMR (400 MHz, DMSO-d6) δH 8.27 (2H, d, j = 8.6 Hz, 2ArH), 8.10 (2H, d, j = 7.8 Hz, 2ArH), 7.70 (1H, t, j = 7.3 Hz, ArH), 7.66 - 7.58 (2H, m, 2ArH), 7.29 (1H, s, =CH), 7.10 (2H, d, j = 8.6 Hz, 2ArH) (Figure S1).
13C NMR (126 MHz, DMSO-d6) δC 174.2, 170.8, 159.3, 157.6, 131.9, 129.6, 128.8, 127.8, 125.7, 123.9, 121.5, 119.9, 116.4, 114.5, 55.8, 34.4 (Figure S2).
HRMS (ESI): m/z [M+Na]+ calcd for C17H13NNaO3: 302.2842 found: 302.2848
2.3.2. (Z)-4-((2-hydroxynaphthalen-1-yl)methylene)- 2-phenyloxazol-5(4H)-one 1h
Following GP1 using benzoylglycine (179 mg, 1 mmol) and 2-hydroxy-1-naphthaldehyde (172 mg, 1 mmol) gave 1h as a pale-yellow solid; yield: 22 mg (7%); m.p. 171.6-172.3 °C.
1H NMR (400 MHz, DMSO-d6) δH 8.88 (1H, d, j = 8.1 Hz, ArH), 8.84 (1H, d, j = 8.1 Hz, ArH), 8.79 (1H, d, j = 8.1 Hz, ArH), 8.37 (1H, s, ArH), 8.20 (1H, t, j = 7.7 Hz, ArH), 8.11 (1H, d, j = 4.8 Hz, 1H, ArH), 8.08 (1H, d, j = 7.7 Hz, ArH), 7.87 (1H, m, ArH), 7.82 (1H, d, j = 5.4 Hz, ArH), 7.58 (1H, d, j = 6.6 Hz, ArH), 7.54 (1H, d, j = 5.6 Hz, ArH), 7.33 (1H, t, j = 6.5 Hz, =CH) (Figure S3).
13C NMR (126 MHz, DMSO-d6) δC 170.9, 165.2, 154.4, 136.2, 132.9, 130.7, 129.1, 128.1, 127.8, 123.6, 122.2, 122.1, 117.2, 71.3, 41.9, 40.5 (Figure S4).
HRMS (ESI): m/z [M+H]+ calcd for C20H14NO3: 316.0968 found: 316.0972
2.3.3. (Z)-4-(4-nitrobenzylidene)-2-phenyloxazol -5(4H)-one 1i
Following GP1 using benzoylglycine (179 mg, 1 mmol) and 4-nitrobenzaldehyde (151 mg, 1 mmol) gave 1i as a yellow solid; yield: 232 mg (79%); 161.3-161.9 °C.
1H NMR (400 MHz, DMSO-d6) δH 8.34 (1H d, j = 8.6 Hz, ArH), 8.15 (1H, d, j = 8.6 Hz, ArH), 7.95 (1H, d, j = 9.1 Hz, 2ArH), 7.74 (1H, d, j = 8.6 Hz, ArH), 7.63 (1H, d, j = 8.6 Hz, ArH), 7.42 (1H, s, ArH), 6.72 (1H, s, =CH), 6.60 (1H, d, j = 9.2 Hz, ArH) (Figure S5).
13C NMR (126 MHz, DMSO-d6) δC 174.6, 169.0, 147.4, 137.1, 133.4, 133.3, 131.9, 131.6, 130.8, 129.5, 129.0, 129.0, 127.3, 117.7, 113.7, 53.4 (Figure S6).
HRMS (ESI): m/z [M+H]+ calcd for C16H11N2O4: 295.0713 found: 295.0715, m/z [M+Na]+ calcd for C16H10N2NaO4: 317.0533 found: 317.0539
2.3.4. (Z)-2-(3,4-dimethoxyphenyl)-4-(furan-2-ylmethylene)oxazol-5(4H)-one 1j
Following GP1 using (3,4-dimethoxybenzoyl)glycine (239 mg, 1 mmol) and furan-2-carbaldehyde (96 mg, 1 mmol) gave 1j as a yellow solid; yield: 89 mg (30%); m.p. 142.9-143.5 °C.
1H NMR (400 MHz, DMSO-d6) δH 7.76 (1H, dd, j = 7.5, 1.5 Hz, ArH), 7.62 (1H, dd, j = 7.5, 1.6 Hz, ArH), 7.29 (1H, s, CH), 7.21 (1H, d, j = 1.5 Hz, ArH), 6.98 (1H, d, j = 7.5 Hz, ArH), 6.79 (1H, dd, j = 7.5, 1.0 Hz, ArH), 6.64 (1H, t, j = 7.5 Hz, ArH), 3.86 (6H, s, 2-OCH3) (Figure S7).
13C NMR (126 MHz, DMSO-d6) δC 174.7, 169.1, 152.2, 141.3, 137.1, 133.2, 132.1, 130.8, 129.7, 129.4, 129.1, 126.4, 115.1, 55.7, 31.16, 21.5 (Figure S8).
HRMS (ESI): m/z [M+H]+ calcd for C16H14NO5: 300.0866 found: 300.0872
2.3.5. (Z)-2-(4-methoxyphenyl)-4-(thiophen-2-ylmethylene)oxazol-5(4H)-one 1k
Following GP1 using (4-methoxybenzoyl)glycine (209 mg, 1 mmol) and thiophene-2-carbaldehyde (112 mg, 1 mmol) gave 1k as a yellow solid; yield: 182 mg (64%); m.p. 154.6-154.8 °C.
1H NMR (400 MHz, DMSO-d6) δH 10.34 (1H s, ArH), 8.76 (1H, s, ArH), 8.11 (2H, d, j = 8.0 Hz, 2ArH), 7.84 (1H, s, =CH), 7.83 - 7.80 (1H, m, ArH), 7.56 - 7.49 (2H, m, 2ArH), 4.12 (3H, s, OCH3) (Figure S9).
13C NMR (126 MHz, CDCl3) δC 166.7, 164.4, 153.5, 148.7, 136.9, 133.1, 130.8, 130.5, 129.9, 127.8, 126.7, 123.4, 122.5, 119.1, 63.8 (Figure S10).
HRMS (ESI): m/z [M+Na]+ calcd for C15H11NNaO3: 308.0352 found: 308.0348
2.3.6. (Z)-2-(4-nitrophenyl)-4-(thiophen-2-ylmethylene)oxazol-5(4H)-one 1l
Following GP1 using (4-nitrobenzoyl)glycine (224 mg, 1 mmol) and thiophene-2-carbaldehyde (112 mg, 1 mmol) gave 1l as a yellow solid; yield: 120 mg (40%); m.p. 186.1-186.7 °C.
1H NMR (400 MHz, DMSO-d6) δH 8.32 (2H, d, j = 8.6 Hz, 2ArH), 8.15 (2H, d, j = 8.6 Hz, 2ArH), 7.72 (2H, d, j = 8.6 Hz, 2ArH, 7.60 (2H, d, j = 8.6 Hz, 2ArH), 7.39 (1H, s, =CH) (Figure S11).
13C NMR (126 MHz, CDCl3) δC 167.3, 165.0, 153.5, 148.7, 136.9, 132.0, 129.1, 128.3, 126.6, 126.5, 123.0, 122.4, 119.1 (Figure S12).
HRMS (ESI): m/z [M+H]+ calcd for C14H9N2O4S: 301.0278 found: 301.0282
2.3.7. (Z)-4-((2-hydroxynaphthalen-1-yl)methylene) -2-(thiophen-2-yl)oxazol-5(4H)-one 1m
Following GP1 using (thiophene-2-carbonyl)glycine (185 mg, 1 mmol) and 2-hydroxy-1-naphthaldehyde (172 mg, 1 mmol) gave 1m as a yellow solid; yield: 135 mg (42%); m.p. 201.5-202.3 °C.
1H NMR (400 MHz, DMSO-d6) δH 10.29 (1H, s, OH), 7.68 (1H, s, ArH), 7.63 - 7.55 (1H, m, ArH), 7.45 - 7.35 (1H, m, ArH), 7.35 - 7.33 (1H, m, ArH), 7.32 (2H, d, j = 2.3 Hz, 2ArH), 7.31 - 7.28 (2H, m, 2ArH), 7.28 - 7.23 (1H, m, ArH), 6.99 - 6.94 (1H, m, ArH), 6.93 - 6.88 (1H, m, =CH) (Figure S13).
13C NMR (126 MHz, DMSO-d6) δC 166.4, 163.9, 148.3, 145.8, 136.1, 128.2, 126.5, 125.5, 124.3, 123.3, 121.7, 120.9, 119.2, 116.5, 113.6, 61.5, 56.4 (Figure S14).
HRMS (ESI): m/z [M+H]+ calcd for C18H12NO3S: 322.0532 found: 322.0536
2.3.8. (Z)-2-(2,4-dichlorophenyl)-4-(4-methoxy benzylidene)oxazol-5(4H)-one 1n
Following GP1 using (2,4-dichlorobenzoyl)glycine (248 mg, 1 mmol) and 4-methoxybenzaldehyde (136 mg, 1 mmol) gave 1n as a yellow solid; yield: 139 mg (40%); m.p. 175.7-176.4 °C.
1H NMR (400 MHz, DMSO-d6) δH 9.53 (1H, d, j = 8.0 Hz, ArH), 7.56 (4H, dd, j = 12.2, 3.3 Hz, 4ArH), 6.74 (2H, d, j = 8.9 Hz, 2ArH), 6.59 (1H, dd, J = 15.6, 8.0 Hz, CH) (Figure S15).
13C NMR (126 MHz, DMSO-d6) δC 173.8, 170.4, 157.6, 133.1, 132.0, 131.9, 129.6, 129.3, 129.1, 129.1, 128.2, 123.6, 121.4, 121.1, 119.9, 116.4, 34.5 (Figure S16).
HRMS (ESI): m/z [M+Na]+ calcd for C17H11Cl2NNaO3: 370.0008 found: 370.0004
2.3.9. (Z)-2-(2-fluorophenyl)-4-(4-methoxybenzyli dene)oxazol-5(4H)-one 1o
Following GP1 using (2-fluorobenzoyl)glycine (197 mg, 1 mmol) and 4-methoxybenzaldehyde (136 mg, 1 mmol) gave 1o as a yellow solid; yield: 133 mg (45%); m.p. 188.2-188.8 °C.
1H NMR (400 MHz, DMSO-d6) δH 8.40 (1H, d, j = 1.9 Hz, ArH), 8.17 (1H, dd, j = 8.8, 1.9 Hz, ArH), 8.05 (1H, d, j = 5.0 Hz, ArH), 7.82 (1H, d, j = 3.5 Hz, ArH), 7.70 (1H, d, j = 16.2 Hz, ArH), 7.64 (1H, s, =CH), 7.44 (1H, d, j = 8.9 Hz, ArH), 7.28 - 7.21 (1H, m, ArH), 7.15 (1H, d, J = 16.2 Hz, ArH), 3.99 (3H, s, OCH3) (Figure S17).
13C NMR (126 MHz, DMSO-d6) δC 173.8, 170.3, 163.1, 161.2, 157.2, 134.6 (d, J = 10.6 Hz), 132.1, 130.9 (d, J = 9.0 Hz), 129.7, 128.3, 123.8, 123.5, 121.4, 120.0, 115.6 (d, J = 20.7 Hz), 114.8, 34.5 (Figure S18).
HRMS (ESI): m/z [M+H]+ calcd for C17H13FNO3: 298.0874 found: 298.0878
2.3.10. (Z)-2-cyclopropyl-4-(4-methoxybenzylidene) oxazol-5(4H)-one 1p
Following GP1 using (cyclopropanecarbonyl)glycine (143 mg, 1 mmol) and 4-methoxybenzaldehyde (136 mg, 1 mmol) gave 1p as a yellow solid; yield: 104 mg (43%); m.p. 128.2-128.5 °C.
1H NMR (400 MHz, DMSO-d6) δH 8.77 (1H, s, ArH), 8.12 (1H, d, j = 8.0 Hz, ArH), 7.85 - 7.82 (2H, m, 2ArH), 7.56 - 7.50 (1H, m, CH), 4.18 (1H, q, j = 7.1 Hz, =CH), 4.12 (3H, s, OCH3), 1.86 - 1.71 (1H, m, CHH), 1.67 - 1.52 (1H, m, CHH), 1.25 (2H, dt, j = 14.2, 5.9 Hz, CH2) (Figure S19).
13C NMR (126 MHz, DMSO-d6) δC 170.3, 164.8, 154.9, 143.1, 138.6, 132.1, 129.4, 129.1, 127.5, 126.1, 125.8, 120.7, 118.1, 115.3, 71.1, 21.7, 18.9 (Figure S20).
HRMS (ESI): m/z [M+H]+ calcd for C17H13FNO3: 266.0788 found: 266.0782
2.3.11. (Z)-2-((E)-3-methoxy-4-nitrostyryl)-4-(thiophen-2-ylmethylene)oxazol-5(4H)-one 1q
Following GP1 using (E)-(3-(3-methoxy-4-nitrophenyl) acryloyl)glycine (280 mg, 1 mmol) and thiophene-2-carbaldehyde (112 mg, 1 mmol) gave 1q as a yellow solid; yield: 121 mg (34%); m.p. 211.2-211.5 °C with decomposition.
1H NMR (400 MHz, DMSO-d6) δH 8.40 (1H, d, j = 1.9 Hz, ArH), 8.17 (1H, dd, j = 8.8, 1.9 Hz, ArH), 8.05 (1H, d, j = 5.0 Hz, ArH), 7.82 (1H, d, j = 3.5 Hz, ArH), 7.70 (1H, d, j = 16.2 Hz, =CH), 7.64 (1H, s, =CH), 7.44 (1H, d, j = 8.9 Hz, ArH), 7.27 - 7.22 (1H, m, ArH), 7.15 (1H, d, J = 16.2 Hz, =CH), 3.99 (3H, s, OCH3) (Figure S21).
13C NMR (126 MHz, DMSO-d6) δC 173.8, 168.6, 138.7, 137.8, 136.0, 135.6, 133.1, 132.0, 130.0, 129.9, 129.1, 127.1, 126.9, 126.5, 112.3, 52.8, 21.2 (Figure S22).
HRMS (ESI): m/z [M+Na]+ calcd for C17H12N2NaO5S: 379.3412 found: 379.3422
2.3.12. 2-((E)-4-nitrostyryl)-4-((Z)-3,4,5-trimethoxy benzylidene)oxazol-5(4H)-one 1r
Following GP1 using (E)-(3-(4-nitrophenyl)acryloyl) glycine (250 mg, 1 mmol) and 3,4,5-trimethoxybenzal dehyde (196 mg, 1 mmol) gave 1r as a yellow solid; yield: 49 mg (12%); m.p. 191.3-191.5 °C.
1H NMR (400 MHz, DMSO-d6) δH 8.10 (2H, d, j = 8.5 Hz, 2ArH), 8.02 - 7.69 (1H, m, =CH), 7.61 (1H, s, =CH), 7.59 (2H, s, 2ArH), 7.47 (1H, d, j = 2.2 Hz, ArH), 7.45 - 7.38 (1H, m, ArH), 6.92 (1H, d, J = 8.7 Hz, =CH), 3.33 (6H, s, 2OCH3), 2.63 (3H, s, OCH3) (Figure S23).
13C NMR (126 MHz, CDCl3) δC 163.84, 162.63, 160.66, 159.05, 148.71, 141.83, 131.42, 130.30, 129.02, 128.70, 128.30, 126.04, 122.70, 121.41, 120.26, 116.02, 115.84, 115.63, 97.48, 22.71, 14.14 (Figure S24).
HRMS (ESI): m/z [M+H]+ calcd for C21H19N2O7: 411.1187 found: 411.1173
2.3.13. 2-((E)-4-(tert-butyl)styryl)-4-((Z)-4-methoxy benzylidene)oxazol-5(4H)-one 1s
Following GP1 using (E)-(3-(4-(tert-butyl)phenyl) acryloyl)glycine (261 mg, 1 mmol) and 4-methoxybenzal dehyde (136 mg, 1 mmol) gave 1s as a yellow solid; yield: 104 mg (7%); m.p. 128.2-128.5 °C.
1H NMR (400 MHz, DMSO-d6) δ 7.93 (2H, d, j = 7.4 Hz, 2ArH), 7.52 (1H, s, ArH), 7.50 (1H, d, j = 7.7 Hz, ArH), 7.47 (4H, d, j = 7.9 Hz, 4ArH), 7.14 (1H, d, j = 15.0 Hz, =CH), 6.94 (1H, d, j = 15.1 Hz, =CH), 6.92 (1H, dd, J = 7.7, 1.5 Hz, 2H, 2ArH), 3.84 (3H, s, OCH3), 1.34 (9H, s, 3CH3) (Figure S25).
13C NMR (101 MHz, DMSO-d6) δC 174.03, 169.97, 155.24, 140.17, 138.72, 133.17, 130.38, 129.26, 128.63, 127.67, 127.41, 122.38, 118.14, 114.67, 71.5, 35.09, 21.38, 20.44 (Figure S26).
HRMS (ESI): m/z [M+H]+ calcd for C23H24NO3: 362.1751 found: 362.1749
2.4. Colorimetric Detection of TrxR Activity
TrxR activity was determined using a lysate of A549 human lung carcinoma cells. The cells were homogenized on ice in phosphate-buffered saline (PBS, pH 7.4) containing 1 mM EDTA and a protease inhibitor cocktail (Beijing Solarbio Science & Technology Co., Ltd.) using an Ultra Turrax T25 disperser (Ikawerk, Janke and Kunkel Inc., Staufen, Germany). The resulting lysates were centrifuged at 15,000 × g for 15 min at 4 °C. The protein concentration in the supernatants was determined using the Bradford method.
The assay utilized 5,5'-dithio-bis-(2-nitrobenzoic acid) (DTNB, Sigma-Aldrich, USA) as a substrate. The reaction was performed in a 96-well plate, containing 50 µg of lysate protein, 50 mM potassium phosphate buffer (pH 7.4), 1 mM EDTA, 50 mM KCl, 0.2 mg/mL bovine serum albumin, and 0.25 mM NADPH (Acros Organics, USA). Inhibitors (5 µL) were added, bringing the final volume to 190 µL. The mixture was incubated for 15 min at 24 °C, after which 10 µL of DTNB was added to a final concentration of 0.5 mM.
The change in TrxR activity was measured by monitoring the absorbance at 412 nm for 20 min using a Clariostar plate reader. The background, TrxR-independent reduction of DTNB in the cell lysates, was subtracted from each value. Concentration-response data were plotted using GraphPad Prism software (version 9.01) to determine the IC50 values. Aurumthiomalate was used as a standard inhibitor; the half-inhibition concentration curve is given in Figures S27-S34. TrxR inhibition by sodium aurothiomalate (ATM) is shown as a dose-response curve in the electronic supporting information.
2.5. Colorimetric Detection of GR Activity
The working solution of the reagent was prepared according to the following protocol:
Assay buffer with the following composition: 0.1 M sodium phosphate (pH 7.4) with 5 mM EDTA, 0.1 M Tris (pH 7.6) with 5 mM EDTA, and 1 mg/mL BSA.
Standard GSSG (glutathione dimer): 0.5 mL of 5 mM in assay buffer.
DTNB: 1 mL of 5 mM in assay buffer.
NADPH: 5 mg dissolved in 2 mL of assay buffer.
GSSG: 5 mg dissolved in 2 mL of assay buffer.
Positive control: 50 µg of GR (lyophilized) dissolved in 1 mL of assay buffer.
Cells were homogenized on ice in 0.1-0.2 mL of cold assay buffer and centrifuged at 10,000 × g for 15 min at 4°C. The supernatant was collected for the assay and stored on ice.
Pre-treated samples (2-50 μL) were placed into a 96-well plate, and the volume was brought to 50 μL with assay buffer.
A TNB standard curve was constructed as follows: 0, 2, 4, 6, 8, and 10 μL of the TNB standard were added to a 96-well plate in duplicate to generate 0, 10, 20, 30, 40, and 50 nmol/well standards. The final volume was brought to 100 μL with assay buffer.
GR activity in the samples was determined using a reaction mix. For each well, a total of 50 μL of reaction mix was prepared, consisting of: 40 μL of GR assay buffer, 2 μL of DTNB solution, 2 μL of NADPH solution, and 6 μL of GSSG solution. Then, 50 μL of the reaction mix was added to each test sample and mixed well.
For samples with inhibitor, the corresponding oxazol-5(4H)-one was added (5 μL) to achieve a final concentration of 25 mM.
Optical density at 405 nm was measured immediately over a 25-min period to generate a kinetic curve, and the initial velocity was calculated.
2.6. Cell Viability Assessment by MTT Assay
The cytotoxic effects of the investigated compounds were evaluated on A549 (ATCC CCL-185™) and HEK293T (ATCC CRL-3216™) cell lines using the colorimetric MTT assay. Cells were seeded in 96-well plates (Eppendorf, Germany) at a density of 1 × 103 cells per well and maintained in complete growth medium consisting of DMEM (high glucose, Capricorn Scientific, Germany) supplemented with 10% fetal bovine serum (Capricorn Scientific, Germany), 2 mM L-glutamine (Gibco, USA), 1 mM sodium pyruvate (Gibco, USA), and 1% penicillin/ streptomycin (Capricorn Scientific, Germany). Cultures were incubated under standard conditions at 37 °C in a humidified atmosphere of 5% CO2 for 24 h to allow cell attachment and to reach approximately 70% confluence. Subsequently, the growth medium was carefully aspirated and replaced with fresh medium containing the test compounds at specified concentrations, and the cells were cultured for an additional 72 h.
Following the 24 h attachment period, the culture medium was replaced with fresh medium containing serial dilutions of the test compounds (1i, 1o, 1s). The compounds were added at a volume of 5 µL per well, with final concentrations ranging from 5 to 250 µM. The final DMSO concentration was maintained constant at 2.5% in all treated wells. A control solution containing 2.5% DMSO in culture medium was used as a negative control. After 72 h of exposure, 20 µL of 0.5% MTT solution (HiMedia, India) was added to each well, and the plates were incubated for an additional 3 h to allow formazan crystal formation. The resulting formazan crystals were solubilized, and the absorbance was measured at 570 nm using a CLARIOstar microplate reader (BMG Labtech, Germany).
To ensure statistical robustness, the experiment was performed in three independent biological replicates, each with eight technical replicates per concentration. Taxol was used as a positive control, while vehicle (DMSO) treatment served as the negative control (100% viability). Absorbance data were normalized to the negative control to calculate the percentage of cell viability, with the lowest value set to 0%. Dose-response curves were generated, and half-maximal inhibitory concentration (IC50) values were determined using non-linear regression analysis in GraphPad Prism software (San Diego, USA).
2.7. Western Blot Analysis of TrxR1
Whole-cell protein lysates were prepared from HeLa and SH-SY5Y cell lines to assess TrxR1 protein expression. The proteins were resolved by SDS-PAGE on a 10-14% gradient gel and subsequently electrophoretically transferred onto a polyvinylidene difluoride (PVDF) membrane (Sigma-Aldrich). For immunodetection, the membrane was probed with a primary polyclonal antibody against TrxR1 (Cloud-Clone Corp., Texas, USA). Following incubation with a species-specific horseradish peroxidase (HRP)-conjugated secondary antibody, the immunocomplexes were visualized using a chemiluminescence substrate, luminol (5-amino-2,3-dihydrophthalazine-1,4-dione).
To ensure equal protein loading and transfer efficiency across lanes, the membrane was re-probed with an antibody targeting β-actin, which served as an internal loading control. Full, uncropped images of all blots are available in the electronic supplementary information. To view the original blot images, including molecular weight markers and β-actin controls (Fig. S27). Molecular weight marker for HeLa cell line; (Fig. S28). Molecular weight marker for SH-SY5Y cell line; (Fig. S29). β-actin blotting for SH-SY5Y cell line; (Fig. S30). β-actin blotting for the HeLa cell line; (Fig. S31). β-actin blotting for U-251 cell line; (Fig. S32). TrxR1 blotting image for HeLa cell line; (Fig. S33). TrxR1 blotting image for the SH-SY5Y cell line.
2.8. Mathematical Processing of Results
All experimental data were obtained in triplicate to account for the influence of random error and to determine the confidence interval. The data were processed using GraphPad Prism 10.0 software using the appropriate presets for calculating Michaelis-Menten kinetics, inhibition parameters, and for generating histograms. Statistical analysis was performed in GraphPad Prism using the Kruskal-Wallis test, which revealed statistically significant differences between the compared groups. Subsequently, pairwise comparisons were performed using the Mann-Whitney test.
2.9. In Silico Studies
2.9.1. Protein Structure Preparation
The three-dimensional structure of human thioredoxin reductase 1 (TrxR1) was retrieved from the RCSB Protein Data Bank (PDB ID: 3EAN) [26]. The initial protein structure was curated by removing all non-essential components, including heteroatoms, co-crystallized ligands, and water molecules. Subsequently, hydrogen atoms were added to the protein framework, and the system was optimized by retaining only polar hydrogens. This entire preparatory procedure was conducted using the Protein Preparation Wizard within the Schrödinger software suite (Schrödinger, LLC: New York, NY, 2017) [27, 28].
2.9.2. Ligand Library Preparation
A chemical library comprising compounds featuring an α, β-unsaturated carbonyl moiety was subjected to computational preparation. Using the LigPrep module, each compound's geometry was optimized with the MMFFs force field, and potential ionization states were generated to model physiological conditions (pH 7.0 ± 2.0). The resulting ensemble of three-dimensional structures was subsequently utilized for molecular docking simulations employing a standard precision (SP) protocol.
2.9.3. Molecular Docking Protocol
Molecular docking studies were performed to explore the binding interactions within the catalytic site of TrxR1. A defined receptor grid, with dimensions of 20 × 20 × 20 Å and a grid spacing of 0.375 Å, was centered on the selenocysteine residue (Sec498) present in the C-terminal active site. All docking calculations were executed using the Glide module integrated into the Schrödinger software suite.
3. RESULTS
3.1. Rational Design of oxazol-5(4h)-ones
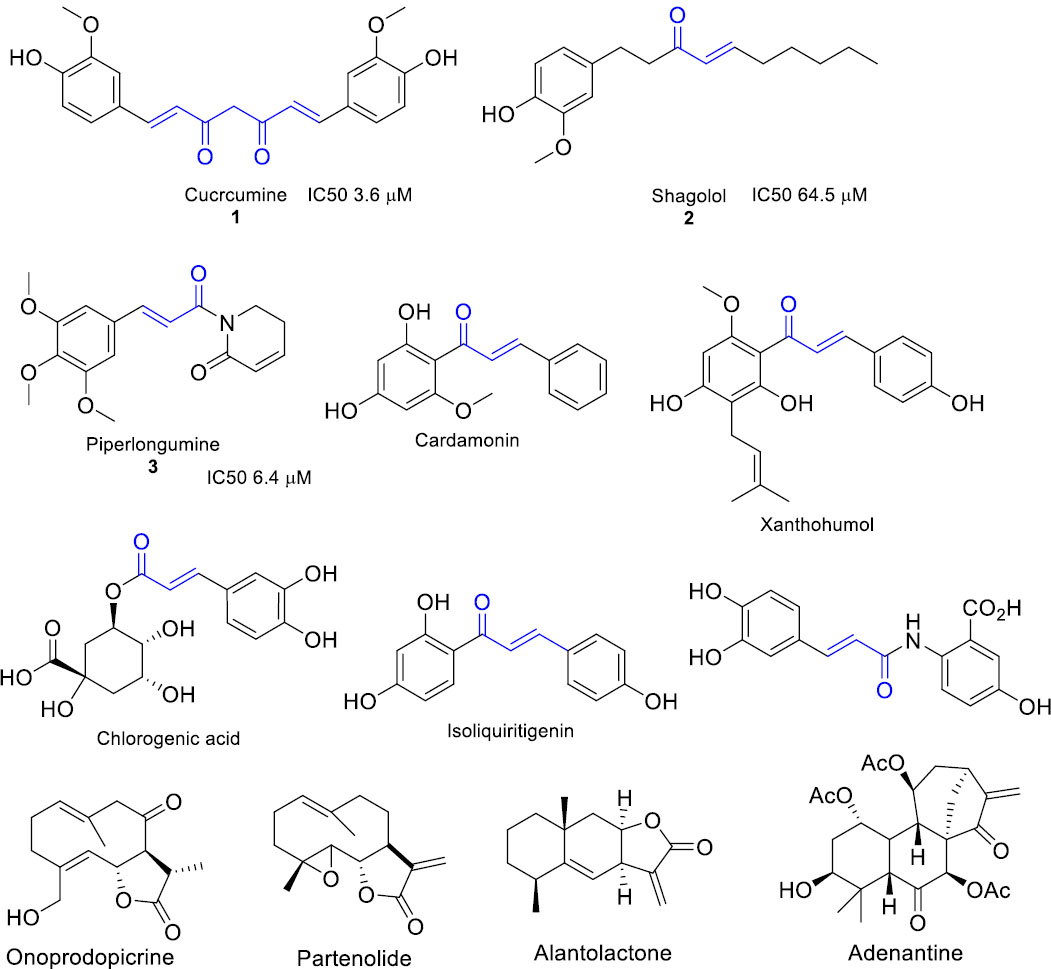
In the first stage of the authors experiments, we used the following oxazol-5(4H)-ones (Fig. 2A) available from the commercial library of ChemDiv (https://www.chemdiv.com). For these compounds, authors determined the level of TrxR1 inhibition using the DTB assay (Fig. 2B). For compounds 1a and 1e, we observed a more than 20% decrease in enzyme activity at a concentration of 25 μM (Fig. 2C). Building on these findings, we conducted a structure-property relationship analysis.
Study of enzyme kinetic TrxR1 for commercial available oxazol-5(H)-ones at 25 µM. A - structure of compounds, B - kinetic curve for TrxR1 in the presence of inhibitors, C - inhibition level for commercial available oxazol-5(H)-ones.
Molecular docking was employed to elucidate the binding poses of target oxazolones within the enzyme's active site and to rationally guide the selection of substituents for the planned syntheses from hippuric acids and corresponding aldehydes. Given prior observations correlating substituent electronic effects with inhibitory activity, the authors analysis for the oxazolone series specifically focused on key quantum-chemical descriptors: the energy of the lowest unoccupied molecular orbital (LUMO), reflecting the compound's electron-accepting capability, and the Mulliken charge on the electrophilic olefinic carbon atom. The calculated values of molecular descriptors were 1a LUMO energy have - 2.4 eV, ovality 1.47, area 315.49 Å2, 1e LUMO energy have -1.2 eV, 1f LUMO energy have +0.9 eV, ovality 1.47, area 311.77 Å2. Thus, our analysis of molecular descriptors for active TrxR1 inhibitors indicates that promising candidates are characterized by lower molecular surface area and ovality values, alongside a lowest unoccupied molecular orbital (LUMO) energy within the range of approximately -2.5 to -1 eV. For instance, compound 1a, with a LUMO energy of -2.4 eV, an area of 315.49 Å2, and an ovality of 1.47, aligns well with these parameters, whereas compounds such as 1f (+0.9 eV) exhibit LUMO energies outside the optimal range. These criteria can serve as useful guidelines for the selection and design of new oxazol-(5H)-one-based inhibitors with enhanced binding potential and optimized electronic properties.
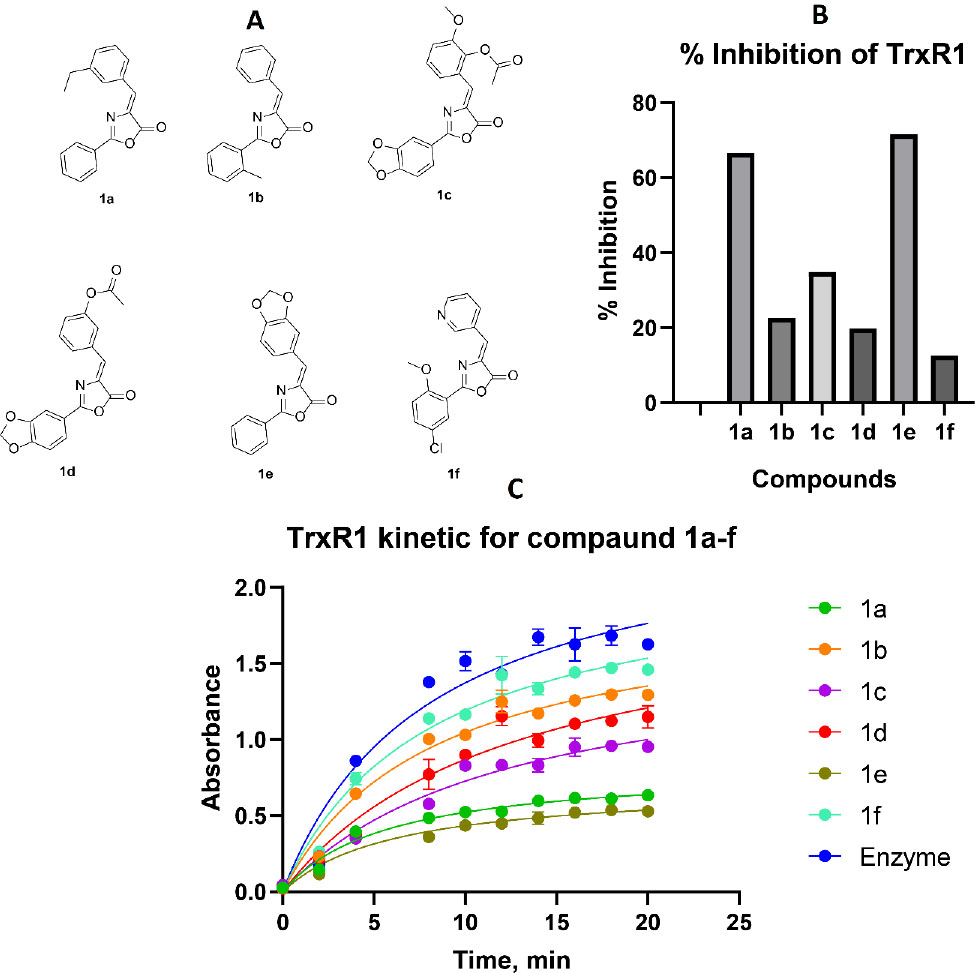
The results obtained inspired authors to conduct a molecular docking analysis (Fig. 3) of the binding of this series of compounds to thioredoxin reductase to evaluate the pharmacophore model. To gain deeper insight into the binding modes of the most active inhibitors within the thioredoxin reductase active site, we performed additional molecular docking simulations. Ligand modeling was carried out using the Glide algorithm, followed by binding affinity estimation via the MM-GBSA model. This analysis revealed that a stable active conformation of the inhibitor is significantly stabilized by key hydrogen bonds between hydrogen bond acceptors in the inhibitor and specific donor residues in the active site-namely, the hydroxyl group of Tyr480, the peptide backbone of Gly470, and the selenol group of the catalytic Sec498.
Ligand docking and interaction of the most active oxazolone 1a and 1e from the ChemDiv library in the active enzyme site. A - ligand interaction diagram and binding pose for 1a, B - ligand interaction diagram and binding pose for 1e.
Molecular docking studies of lead compound 1a ((Z)-4-(3-ethylbenzylidene)-2-phenyloxazol-5(4H)-one) against human TrxR1 (PDB: 3EAN) revealed a well-defined pharmacophore essential for inhibition (Fig. 2). Three critical features govern binding: Electrophilic warhead: The exocyclic C4=C5 olefin positioned 6.85 Å from Sec498 selenol (-SeH), primed for Michael addition. Hydrogen-bond anchor: Oxazolone C2=O formed dual H-bonds with Gly499 backbone NH 3.68 Å. Aryl binding pocket: 4-ethylbenzylidene occupied a hydrophobic cleft bordered by Pro344, Thr343, Glu 477, Ile 478, Thr480 in edge-to-face π-stacking with Thr481. Energy decomposition analysis highlighted key contributions non-covalent interactions contributed -42.9 kcal/mol for 1e (van der Waals: 73%, H-bonding: 27%) and -37.14 kcal/mol for 1a (van der Waals: 81%, H-bonding: 19%). Pharmacophore refinement: We optimized the model by introducing weak EWG substituents to enhance electrophilicity and hydrophobicity for greater contribution of the van der Waals interaction.
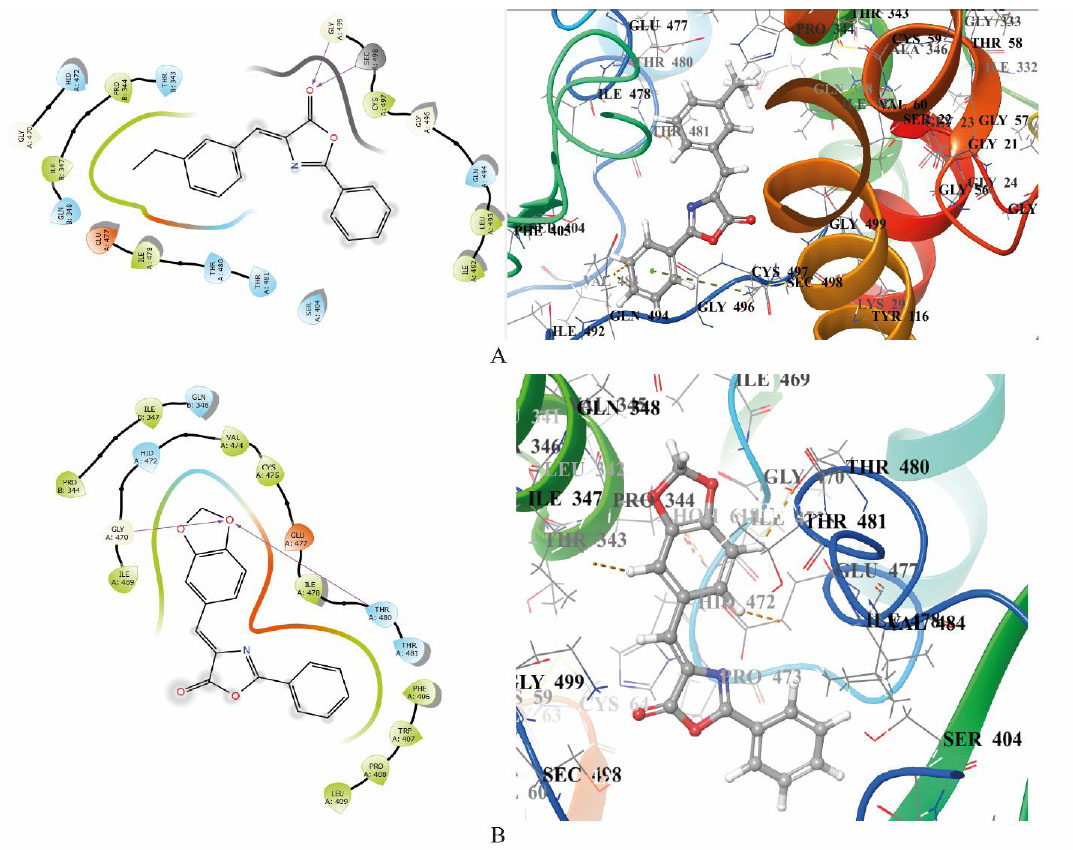
Based on the results of molecular docking and pharmacophoric modeling, as well as our early QSAR work [31], the authors selected the following structures for the targeted synthesis of a pilot series of oxazol-5(4H)ones (Scheme 1) to test the hypothesis about the possibility of creating effective inhibitors of TrxR1.
Synthesis of series of new oxazol-5(4H)-ones.
The target compounds were synthesized in a multicomponent format, simultaneously mixing the corresponding hippuric acids and aldehydes under dehydration conditions with anhydrous sodium acetate and propionic anhydride. The yields of the target oxazolones were average. It is worth noting that compounds 1q - s were obtained from the corresponding glycine amide and cinnamic acid; such dual michael acceptors ensured the formation of covalent dimers of TrxR1, which will be discussed below.
3.2. Studies of Inhibition TrxR1 and GR by oxazol-5(4H)-ones
The obtained compounds were tested as inhibitors of TrxR1 using kinetic analysis with DTNB on lysates of lung carcinoma cells (A549) at a concentration of 25 µM (Fig. 4). Based on the kinetic data, graphs were plotted, and the enzymatic reaction rates were determined relative to the slope to the X-axis. A lysate with 5 µL of DMSO was used as a negative control, while a solution of aurothiomalate (20 mM) served as a positive control. To assess the cross-reactivity of the molecules, an analysis of glutathione reductase (GR) inhibition was conducted, as both enzymes share a similar structure and play a role in redox balance. Since glutathione reductase maintains glutathione levels, its inhibition may lead to increased oxidative stress, which could be detrimental to healthy cells. The testing will ensure the selectivity of the drug, minimize undesirable effects, and enhance therapeutic safety, which aims to inhibit TrxR1. The figures show the obtained inhibition graphs for thioredoxin reductase, and the table lists the activity values of TrxR1 and GR as percentages relative to the negative control.
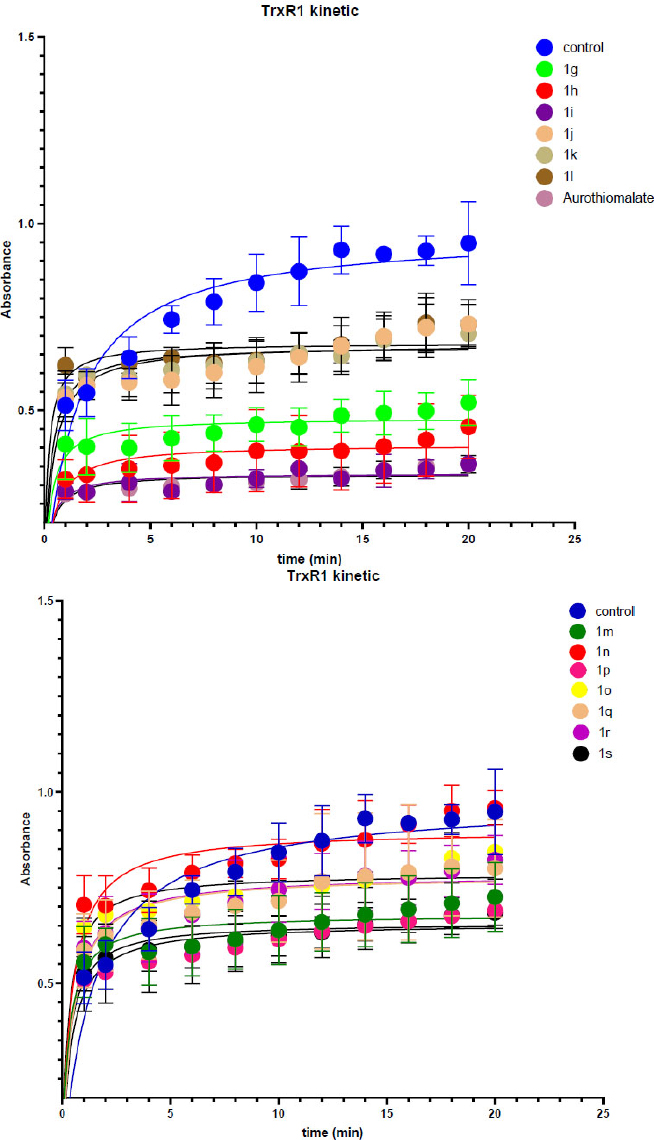
Inhibition graphs of thioredoxin reductase by a series of synthesized oxazol-5(4H)-ones.
Results of the kinetic experiment we used for analysis of activity inhibition for SAR and Lead selection.
3.3. Lead Selection and Structure-Activity Relationships
To select the leading compounds, we constructed a histogram based on the kinetic experiment data. The initial rate of the native enzyme measured without the presence of the inhibitor was taken as 100%; the residual activity was determined as a relative proportion of the native enzyme activity. A histogram of inhibitory activity was constructed for thioredoxin reductase and glutathione reductase. The histogram (Fig. 5) shows the levels of TrxR1 and GR inhibition by a series of oxazol-5(4H)-one compounds.
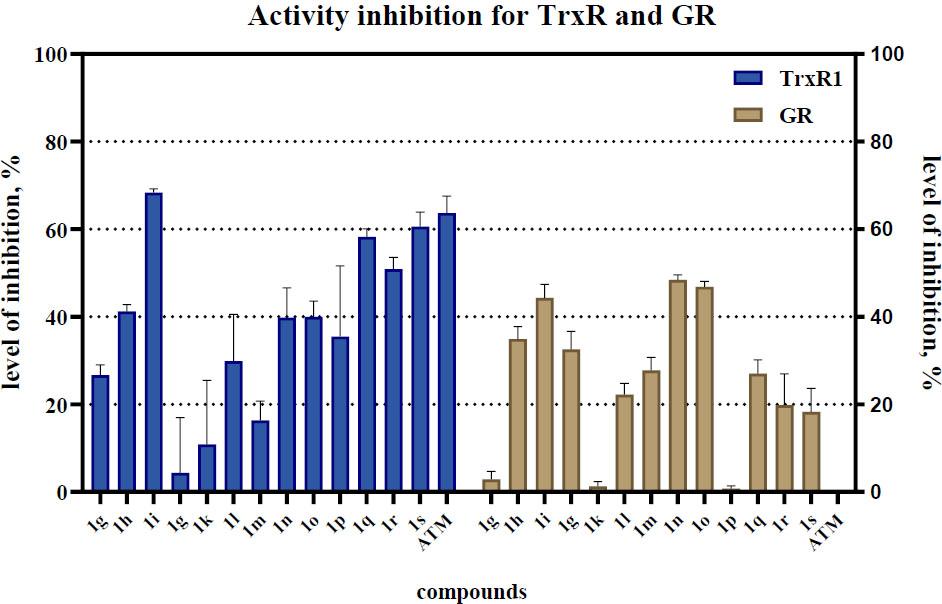
Histogram of levels of activity inhibition for TrxR1 and GR. The three most promising inhibitors of TrxR1 (1i, 1o, and 1s) were identified as lead compounds.
The maximum suppression of enzymatic activity (over 50%) was recorded for compounds 1i, 1o, and 1s with minor off-target activity on GR. Compounds 1j, 1k, and 1m exhibited weak inhibitory activity, reducing the rate of the enzymatic reaction by no more than 20%. Notably, compound 1s surpassed the efficacy of aurothiomalate (the positive control), indicating its high inhibitory potential. Comprehensive profiling of 18 oxazolones at 25 μM (Fig. 4) revealed striking structure-activity relationships: dominant electronic effects correlation: %Inh = 35.7(σₚ*) + 32.1 (R2=0.93), confirming electron deficiency as the primary activity driver. Thiophene analogs (1k, 1l) showed moderate activity (35-48% inh.), with Cβ-thienyl (1l) outperforming Cα-isomers due to improved planarity (dihedral: 8.7° vs 32.5°). Furan-derived 1j exhibited weak inhibition (22.1%), attributable to reduced electrophilicity (LUMO: -1.45 eV) and poorer hydrophobic matching. We have separately noted the design strategy of double Michael acceptors. Compounds with extended conjugation (1q-s) demonstrated divergent behavior: 1q (thienyl + nitrostyryl): 48.9% TrxR1 inhibition but compromised selectivity (GR inh: 51.9%) 1s (styryl + methoxybenzylidene): Balanced profile (TrxR1: 51.7%, GR: 6.9%) with enhanced cellular uptake (logP = 4.1 vs 1i 2.9) Lead selection rationale: 1i prioritized for potency (highest TrxR1 inhibition, IC50 = 25 µM) 1o selected for selectivity (TrxR1/GR ratio: 6.7) 1s advanced for drug-like properties (clogP 4.1, tPSA 78 Å2, ligand efficiency 0.38). While strong electron-withdrawing groups universally enhance enzymatic inhibition, selectivity requires careful modulation of lipophilicity and steric bulk. Compound 1o exemplifies this balance - its ortho-fluorine improves membrane permeation while maintaining low GR cross-reactivity, translating to exceptional tumor selectivity.
3.4. Cellular Activity for Lead Compounds
Analysis of enzymatic activity revealed the selectivity of the synthesized compounds towards TrxR1 compared to GR. Most compounds demonstrated significantly higher inhibitory activity against TrxR1, reducing its reaction rate by more than 65%, likely due to their affinity for the selenium-containing active site of this enzyme. In contrast, glutathione reductase (GR) exhibited considerably lower sensitivity to the studied compounds. Exceptions were 1j and 1n, which showed increased affinity specifically for GR, inhibiting its activity by 51.9% and 48.1%, respectively. Structural analysis indicated that these compounds contain a 4-nitrostyryl-oxazolone fragment, suggesting its key role in interaction with GR. The most potent inhibitor of TrxR1 was compound 1i, which suppressed its activity by more than 68%. Based on the obtained data, promising candidates (1i, 1o, 1s) were selected, reducing the activity of TrxR1 by more than 50%. For these compounds, half-maximal inhibitory concentrations (IC50) were determined within the concentration range of 0.01-20 mM (Fig. 6).
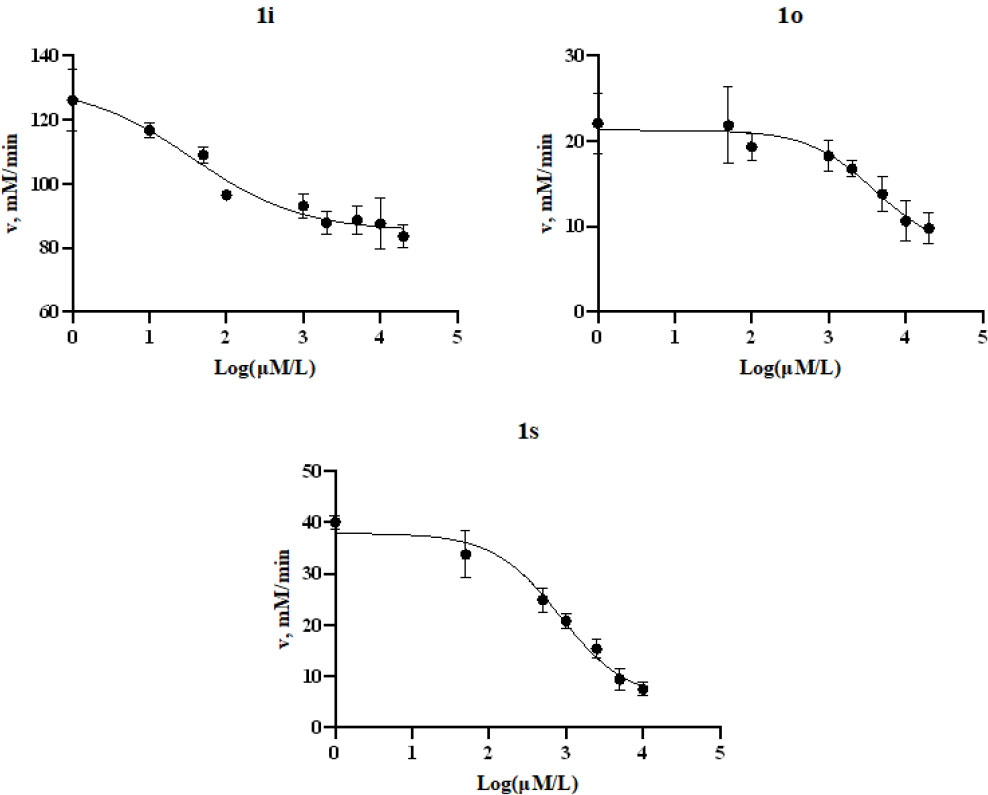
IC50 graph for inhibition TrxR1 oxazol-5(4h)-one for 1i, 1o, 1s.
To determine the cytotoxic effect on cell lines, five cell lines were selected: HEK293T as a healthy cell line, and tumour cell lines of lung carcinoma (A549), neuroblastoma (SHSY5Y), glioblastoma (U251), and cervical cancer (HELA). Studies were conducted over a concentration range of 1-250 µM. Based on the obtained data, dose-response curves were constructed, and the half-maximal cytotoxic concentration (CC50) values were calculated. The results are presented in Table 1.
| - | IC50 inhib. TrxR1, nM | IC50 (A549), μM | IC50 (HEK293T), μM | IC50 (SHSY5Y), μM | IC50 (U251), μM | IC50 (Hela), μM |
|---|---|---|---|---|---|---|
| 1i | 0.25 | 171.3 | 51.89 | 63.53 | 12.58 | 34.86 |
| 1o | 19.7 | 466.1 | 120.1 | 26 | 73.85 | 30.85 |
| 1s | 4.2 | 0,33*10-3 | 0,51*10-3 | 0,31*10-3 | 0,20*10-3 | 0,29*10-3 |
The results revealed different cytotoxicity profiles for the investigated compounds. Compound 1i demonstrated pronounced cytotoxicity against U251 glioblastoma cells (IC50 = 12.58 µM) with a notable 4.12-fold selectivity (SI = 4.12) over normal HEK293T cells (IC50 = 51.89 µM). However, it showed no selectivity against A549 (SI = 0.30) and SHSY5Y (SI = 0.82) cell lines, indicating its effect is line-specific. The selectivity index in Table 2 was present. Compound 1o exhibited the highest selectivity against SHSY5Y neuroblastoma cells (SI = 4.62), while also showing significant activity against HeLa cells (SI = 3.89). Compound 1s showed a consistent, moderate selectivity across all tested tumor lines, with SI values ranging from 1.55 (A549) to 2.55 (U251), confirming its broad-spectrum potential. A clear dose-dependent dynamic of cytotoxic effects was observed for all compounds across all tested cell lines. Analysis of the selectivity indices (SI = IC50 for normal cells / IC50 for tumor cells) highlighted the distinct activity profiles of the compounds (Table 2). Compounds 1i and 1o exhibited narrow, targeted selectivity against specific cell lines (U251 and SHSY5Y/HeLa, respectively). In contrast, compound 1s demonstrated a broad spectrum of moderately selective cytotoxicity, making it the most promising candidate for further investigation as an agent with potential polyvalent activity. The identified selectivity profiles indicate the presence of a potential therapeutic window for further development.
| - | Selectivity Index (SI) | |||
|---|---|---|---|---|
| (HEK293T/A549) | (HEK293T/SHSY5Y) | (HEK293T/U251) | (HEK293T/Hela) | |
| 1i | 0.30 | 0.82 | 4.12 | 1.49 |
| 1o | 0.26 | 4.62 | 1.63 | 3.89 |
| 1s | 1.55 | 1.65 | 2.55 | 1.76 |
To assess changes in the expression level of the target enzyme, immunoblot analysis was conducted on three cell lines that demonstrated the highest cytotoxic response: SHSY5Y (neuroblastoma), U251 (glioblastoma), and HELA (cervical carcinoma). However, in the U251 cell line, detection of the target enzyme proved impossible, which may indicate a lack of its expression in this line. The presented figure (Fig. 7) shows the results of immunoblotting for the two cell lines (SHSY5Y and HELA), where the expression of the investigated enzyme was successfully detected.
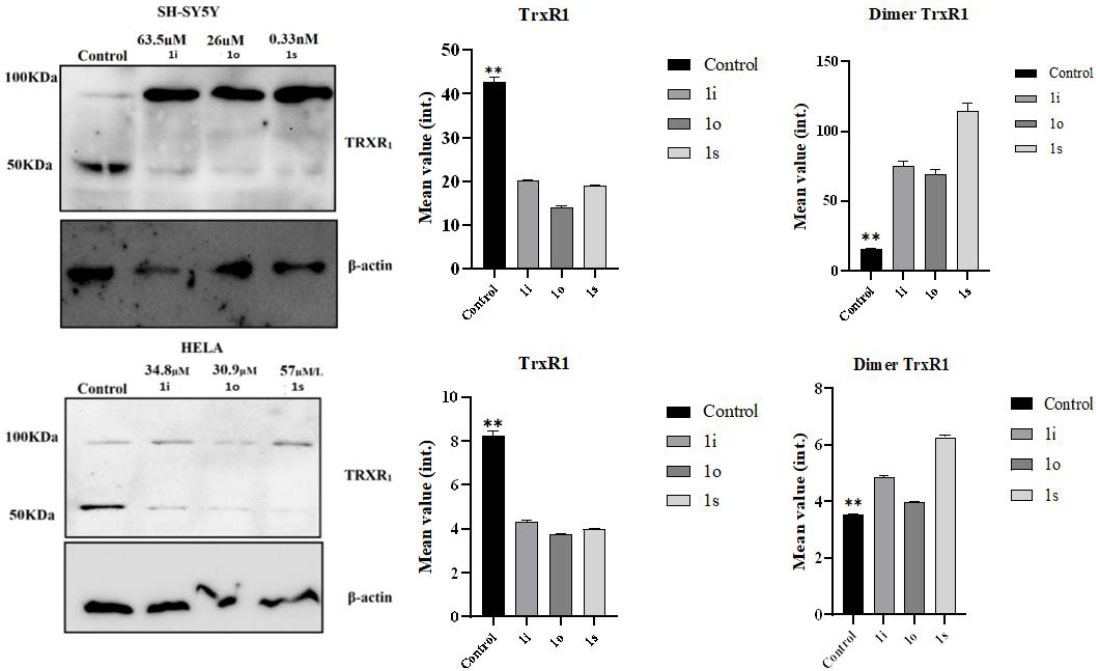
Blotting analysis of TRXR1 expression in SHSY5Y (neuroblastoma) and HELA (cervical carcinoma) cell lysates without and in the presence of lead compounds 1i, o, s. (Mann Whitney test, **- p-value = 0,0022).
(For original blotting data, refer to Figs. S27-S33 ESI) Notably, during the analysis, immunoreactive bands were detected with a molecular weight approximately double that of the calculated mass of the monomeric form of TrxR1 (≈115 kDa compared to ≈55 kDa). This observation aligns with literature data describing the formation of covalent dimers of TrxR1 upon inhibition by electron-deficient olefins [32]. The mechanism of this process is presumably associated with the blocking of the catalytic selenocysteine residue, leading to intermolecular crosslinking. The highest intensity of the dimer signal was observed in SHSY5Y cells after treatment with compound 16I (a fivefold increase compared to the control). In HeLa cells, the dimeric form was also detected, but with lower intensity (2-3 times increase relative to the control). It should be noted that a weak dimer signal was also detected in untreated cells, which may reflect the basal level of enzyme dimerization in vivo.
4. DISCUSSION
The results of an extensive rational design study of thioredoxin reductase inhibitors, initiated as an investigation of the thioredoxin reductase inhibitory activity of oxazolones obtained from the commercial ChemDiv library, led us to the directed design of a series of new oxazol-5(4H)-one derivatives. The structural design was preliminarily performed taking into account the results of molecular docking of a commercial library of compounds from the ChemDiv collection, for which inhibition levels at a concentration of 25 μM were obtained. We noted that the carbon framework of the oxazolone itself affects the positioning of the molecule in the active site of the enzyme. In this case, a phenyl substituent with weak electron-withdrawing groups should preferably occupy position 2. Based on this, we synthesized 18 compounds with various substituents in a multicomponent format; we would like to draw special attention to compounds 1q, 1r, and 1s, which can be called double Michael acceptors, since they contain two fragments representing electron-deficient olefins.
Such a design led us to the discovery of an interesting effect: the formation of thioredoxin reductase dimers during inhibition, which was confirmed by immunoblotting. This type of inhibition has already been described in the literature [33], and this research demonstrates the possibility of rational design of inhibitors of this type. In our study of thioredoxin reductase inhibition activity, we used sodium aurothiomalate (Figure S34) as a positive control to verify the performance of the biological assay.
This research opens the way to the rational design of thioredoxin reductase inhibitors containing a Michael acceptor as a warhead, with the oxazolone carbon framework acting as a conformational anchor for positioning in the enzyme active site.
The study demonstrates that the synthesized oxazol-5(4H)-one derivatives are highly selective and potent inhibitors of thioredoxin reductase 1 (TrxR1), exhibiting significantly lower activity against glutathione reductase (GR). The exceptions were compounds 1j and 1n, containing a 4-nitrostyryl fragment, indicating a potentially different steric or electronic configuration favorable for interaction with the active site of GR. The observed cross-reactivity of these compounds toward GR led to the discontinuation of their further evaluation, and as a result, they were excluded from the selection of lead compounds.
The most promising TrxR1 inhibitors, compounds 1i, 1o, and 1s, demonstrated nanomolar IC50 values, confirming high affinity for the selenium-containing active site of the enzyme. Importantly, high inhibitory activity against TrxR1 correlated with pronounced cytotoxicity against a number of tumor cell lines. Compound 1i demonstrated the highest efficacy against U251 glioblastoma cells (IC50 = 12.58 µM) with significantly lower toxicity to healthy HEK293T cells (IC50 = 51.89 µM), indicating selectivity and the presence of a therapeutic window (SI = 4.13). Compound 1o demonstrated particularly high selectivity against SH-SY5Y neuroblastoma cells (IC50 ~ 18 µM). Notably, compound 1s demonstrated exceptional activity in the nanomolar range against the U251 cell line. The immunoblotting results partially confirmed the expression of the target enzyme in the sensitive SH-SY5Y and HeLa cell lines. The absence of a detectable TrxR1 signal in the U251 cell line, despite the high cytotoxicity of 1i and 1s, requires further study and may indicate alternative mechanisms of cytotoxic action or metabolic features in these cells. Thus, the obtained data confirm the advisability of further studying oxazolone derivatives as promising compounds for the development of selective antitumor drugs targeting the thioredoxin system.
This study is a logical continuation of early work in the field of thioredoxin reductase inhibitors and represents a completed medicinal-chemical investigation on the early-stage search for biologically active compounds with therapeutic potential. To contextualize the performance of our lead oxazolones, a direct comparison with benchmark TrxR inhibitors is informative. Compound 1i exhibits an enzymatic IC50 of 0.25 nM, which compares favorably to the potent gold-based inhibitor auranofin (IC50 ~ 0.1-1 nM) and significantly outperforms many early-stage covalent inhibitors. While its cellular IC50 values are in the micromolar range-a common feature due to factors such as cellular uptake and metabolism-its selectivity profile against U251 glioblastoma cells (SI = 4.12) is promising. Compound 1o offers exceptional selectivity for TrxR1 over GR (ratio > 6) and notable cell-line specificity (e.g., SI = 4.62 for SH-SY5Y), a valuable trait for reducing off-target toxicity. Compound 1s, despite its puzzling activity in TrxR1-negative U251 cells, shows uniformly moderate selectivity across all tested cancer lines (SI ~ 1.5-2.6) and represents a novel structural archetype. This combination of potent enzymatic inhibition, discernible cellular selectivity, and a novel drug-like scaffold positions these oxazolones as competitive candidates within the landscape of next-generation TrxR inhibitors.
The incorporation of a Michael acceptor pharmacophore, while effective for targeting the nucleophilic Sec498 residue of TrxR1, carries inherent pharmacological risks. Such electrophilic motifs can potentially engage in off-target reactions with other cellular thiols, such as glutathione (GSH) or cysteine residues in non-target proteins, leading to toxicity, rapid conjugation/ detoxification, and unpredictable reactivity. The oxazolone scaffold in our series may mitigate some of these risks. Its compact structure and the ability to fine-tune the electrophilicity of the exocyclic olefin through aryl substituents could allow for a reactivity “sweet spot”-sufficient for targeting TrxR1 but reduced for promiscuous reactions. Furthermore, the scaffold’s overall compliance with drug-likeness rules may improve metabolic stability. Future studies will need to directly assess glutathione conjugation rates and perform broader proteomic profiling to evaluate the selectivity of these compounds in a complex biological milieu.
5. STUDY LIMITATIONS
This work has several limitations that define the scope of current findings and guide future research. First, the cytotoxic evaluation was performed on a limited panel of cancer cell lines. Second, and most notably, the study lacks in vivo pharmacokinetic and efficacy data, which are essential for assessing the therapeutic potential and safety profile of these compounds. Third, while GR was tested, a more comprehensive investigation of off-target effects against other redox-active enzymes or through proteome-wide profiling was not conducted. Finally, the observed potent cytotoxicity in U251 cells lacking detectable TrxR1 expression underscores the possibility of alternative mechanisms or redox network compensation, highlighting the complexity of targeting antioxidant systems in cancer. Addressing these limitations will be the focus of our subsequent research.
CONCLUSION
In conclusion, in this work, the rational design and synthesis of a new series of oxazol-5(4H)-one derivatives aimed at inhibiting thioredoxin reductase 1 (TrxR1), a key enzyme of the tumor cell antioxidant system, were successfully implemented. Based on molecular docking data and analysis of a commercial compound library, a hypothesis was proposed regarding the key role of the electron-deficient olefin fragment (Michael acceptor) and the aromatic substituent at position 2 of the oxazolone core for effective binding to the selenol-containing active site of the enzyme.
The synthesized compounds demonstrated high and selective inhibitory activity against TrxR1, which was confirmed by kinetic studies. The most promising lead compounds (1i, 1o, and 1s) showed micromolar IC50 values and pronounced cytotoxicity against a number of tumor cell lines (A549, SH-SY5Y, U251, HeLa), while maintaining significantly lower toxicity against normal HEK293T cells, indicating the presence of a therapeutic window.
An important discovery was the observation of covalent TrxR1 dimer formation under the action of compounds containing two Michael acceptor fragments (1q-s), which was confirmed by Western blotting. This indicates a unique mechanism of inhibition associated with cross-linking of enzyme subunits.
In summary, the present study not only expands our understanding of the structure-activity relationship for oxazolone-based TrxR1 inhibitors but also provides a new strategy for the rational design of highly active and selective antitumor agents targeting the thioredoxin system. The results provide a basis for further structural optimization and preclinical studies of promising compounds.
DISCLOSURE
NMR, mass spectrometry, spectrophotometry, and X-ray studies were performed at the Research Centre for Magnetic Resonance, the Centre for Chemical Analysis and Materials Research, the Educational Resource Center of Chemistry, and the Centre for X-ray Diffraction Methods of the Saint Petersburg State University Research Park.
AUTHORS’ CONTRIBUTIONS
The authors confirm their contribution to the paper as follows: E.C. and V.Y.: Study conception and design were carried out; V.Y. and A.R.: Data collection was performed; A.Z., A.O., I.M., and O.L.: Analysis and interpretation of results were conducted; I.M.: The draft manuscript was prepared. All authors reviewed the results and approved the final version of the manuscript.
LIST OF ABBREVIATIONS
| TrxR1 | = Thioredoxin Reductase |
| GR | = Glutathione Reductase |
AVAILABILITY OF DATA AND MATERIALS
The data supporting the findings of the article is available in the [Zendo] at [https://zenodo.org/records/ 20280912?token=eyJhbGciOiJIUzUxMiJ9.eyJpZCI6ImZhMzYwNWFjLTk4MTgtNDMwOS05ZjJmLTllNmY3NTZjNGQ2OCIsImRhdGEiOnt9LCJyYW5kb20iOiJlY2I1NDY0NjRkNTM5YjQ3ZTJmZjI1NGFjODBjYjM3MiJ9.g06j9-rLXwhc2BOH8 uA-6JTAOhvSAgb9SL2hlot71eR6TOjU3F5MuA6EhUD_GitEuSVXOCO-0u1IewhYUW6kOA], this dataset for all data in manuscript.
FUNDING
This work was financial supported by the Russian Science Fund (RFS), Russia grant number 24-23-00603.
ACKNOWLEDGEMENTS
Declared none.

